大连理工大学 AIChE Journal | 加速量子化学驱动的性质预测和分子设计的机器学习潜在模型
计算材料学
2025-06-19 08:00
文章摘要
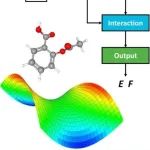
本文介绍了大连理工大学刘奇磊课题组开发的一种基于深度学习的分子势能面预测工具DeePEST,旨在解决量子化学计算成本高的问题。该工具通过引入原子自注意力机制的机器学习势能模型(MLP),显著提高了分子能量和原子力的预测精度和计算效率。DeePEST结合严格的GeomeTRIC优化算法,实现了高效的分子几何优化,并在多个案例研究中展示了其在加速量子化学性质计算、热力学性质预测及计算机辅助分子设计中的广泛应用。与传统量子化学方法相比,DeePEST在保持高精度的同时,计算速度提高了两个数量级以上。未来研究将致力于拓展MLP模型的应用范围,包括过渡态搜索等。
本站注明稿件来源为其他媒体的文/图等稿件均为转载稿,本站转载出于非商业性的教育和科研之目的,并不意味着赞同其观点或证实其内容的真实性。如转载稿涉及版权等问题,请作者速来电或来函联系。
推荐文献

Waterborne transparent anti‐smudge coating with cooling performance via molecular engineering
DOI: 10.1002/aic.70276
Pub Date : 2026-02-06

Issue Editorial Masthead
DOI: 10.1021/efv040i005_2037331
Pub Date : 2026-02-05

Biogenesis of Au Nanoparticles from Plant-Derived Metabolites - <i>In Vitro</i> and <i>In Vivo</i> Studies.
DOI: 10.1021/acsbiomaterials.5c01488
Pub Date : 2026-02-04
计算材料学

佛山大学物理与光电工程学院本科生在一年内连续发表4篇Physical Review B.
2025-12-30
非化学计量钍基碳化物局部晶格畸变与热力学性质研究.
2025-12-30

佛山大学诚聘海内外高层次人才!.
2025-12-30

AI4S回归白盒符号主义,清华等联合发布SR-LLM:自主发现科学知识.
2025-12-30
最新文章





