ACS Catalysis: 分子动力学(MD)模拟+密度泛函理论(DFT)揭示间接配位位点的高效率
计算材料学
2024-10-06 08:00
文章摘要
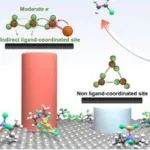
本文通过分子动力学(MD)模拟和密度泛函理论(DFT)研究了乙炔氯化氢化反应中Cu基催化剂的活性位点。研究发现,过量氯化铜诱导的间接配体配位位点具有优越的催化性能,解决了传统Cu基催化剂活性低的问题。实验和理论模拟结果表明,配体增强的Cu基催化剂通过电子转移和链状结构的自发组装,显著提高了催化活性和稳定性。这项工作为合理设计高效的非汞催化剂提供了理论指导,并为复杂催化体系中活性位点的识别提供了新的视角。
本站注明稿件来源为其他媒体的文/图等稿件均为转载稿,本站转载出于非商业性的教育和科研之目的,并不意味着赞同其观点或证实其内容的真实性。如转载稿涉及版权等问题,请作者速来电或来函联系。
推荐文献

Issue Publication Information
DOI: 10.1021/apv008i002_2033340
Pub Date : 2026-01-23

Issue Publication Information
DOI: 10.1021/csv016i003_2037648
Pub Date : 2026-02-06

Atomically Precise Metal Clusters for NIR-II Imaging.
DOI: 10.1021/acs.accounts.5c00837
Pub Date : 2026-02-05
计算材料学

佛山大学物理与光电工程学院本科生在一年内连续发表4篇Physical Review B.
2025-12-30
非化学计量钍基碳化物局部晶格畸变与热力学性质研究.
2025-12-30

佛山大学诚聘海内外高层次人才!.
2025-12-30

AI4S回归白盒符号主义,清华等联合发布SR-LLM:自主发现科学知识.
2025-12-30
最新文章





