{"title":"OXCT1基因的新突变导致琥珀酰辅酶A:3-酮酸辅酶A转移酶(SCOT)缺乏,始于神经系统表现。","authors":"Davoud Amirkashani, Mostafa Asadollahi, Rozita Hosseini, Saeed Talebi, Zahra Golchehre, Mohammad Keramatipour","doi":"10.22037/ijcn.v17i2.35963","DOIUrl":null,"url":null,"abstract":"<p><p>Succinyl-CoA:3-oxoacid CoA-transferase (SCOT) deficiency is an inborn error of ketone body utilization characterized by intermittent ketoacidosis crises. This study reports the first Iranian patient with SCOT deficiency who presented with seizure and hypotonia at birth. Accordingly, she was consequently re-hospitalized due to hypotonia and respiratory distress. Laboratory tests revealed hyperammonemia, ketonuria, and metabolic acidosis. Besides, the plasma glucose level was normal without any other abnormality. Despite treatment with high-dose bicarbonate, severe acidosis persisted. Poor response to treatment raised a significant diagnostic challenge among specialists until genetic investigation identified a homozygous nonsense mutation (c.79G>T; p.Gly27*) in the OXCT1 gene (NM_000436), causing SCOT deficiency. Genetic studies help clinicians achieve a definite diagnosis of such metabolic disorders. In this case, the accurate and early diagnosis of SCOT deficiency opened new therapeutic possibilities, including frequent carbohydrate-rich meals and low fat and protein diet. Moreover, our findings expand the mutational and clinical spectrum of SCOT deficiency<b>.</b></p>","PeriodicalId":14537,"journal":{"name":"Iranian Journal of Child Neurology","volume":"17 2","pages":"127-133"},"PeriodicalIF":0.9000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/94/a3/ijcn-17-127.PMC10114279.pdf","citationCount":"0","resultStr":"{\"title\":\"A Novel Mutation in the OXCT1 Gene Causing Succinyl-CoA:3-Ketoacid CoA Transferase (SCOT) Deficiency Starting with Neurologic Manifestations.\",\"authors\":\"Davoud Amirkashani, Mostafa Asadollahi, Rozita Hosseini, Saeed Talebi, Zahra Golchehre, Mohammad Keramatipour\",\"doi\":\"10.22037/ijcn.v17i2.35963\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Succinyl-CoA:3-oxoacid CoA-transferase (SCOT) deficiency is an inborn error of ketone body utilization characterized by intermittent ketoacidosis crises. This study reports the first Iranian patient with SCOT deficiency who presented with seizure and hypotonia at birth. Accordingly, she was consequently re-hospitalized due to hypotonia and respiratory distress. Laboratory tests revealed hyperammonemia, ketonuria, and metabolic acidosis. Besides, the plasma glucose level was normal without any other abnormality. Despite treatment with high-dose bicarbonate, severe acidosis persisted. Poor response to treatment raised a significant diagnostic challenge among specialists until genetic investigation identified a homozygous nonsense mutation (c.79G>T; p.Gly27*) in the OXCT1 gene (NM_000436), causing SCOT deficiency. Genetic studies help clinicians achieve a definite diagnosis of such metabolic disorders. In this case, the accurate and early diagnosis of SCOT deficiency opened new therapeutic possibilities, including frequent carbohydrate-rich meals and low fat and protein diet. Moreover, our findings expand the mutational and clinical spectrum of SCOT deficiency<b>.</b></p>\",\"PeriodicalId\":14537,\"journal\":{\"name\":\"Iranian Journal of Child Neurology\",\"volume\":\"17 2\",\"pages\":\"127-133\"},\"PeriodicalIF\":0.9000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/94/a3/ijcn-17-127.PMC10114279.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Iranian Journal of Child Neurology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.22037/ijcn.v17i2.35963\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Iranian Journal of Child Neurology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.22037/ijcn.v17i2.35963","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
A Novel Mutation in the OXCT1 Gene Causing Succinyl-CoA:3-Ketoacid CoA Transferase (SCOT) Deficiency Starting with Neurologic Manifestations.
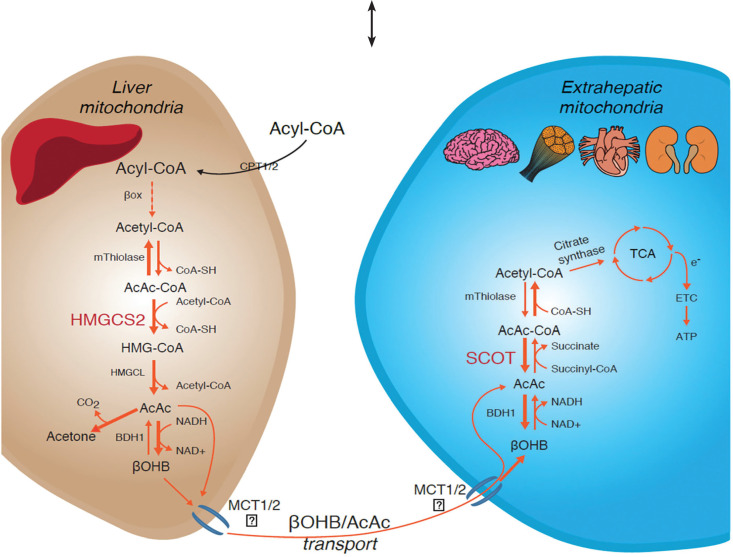
Succinyl-CoA:3-oxoacid CoA-transferase (SCOT) deficiency is an inborn error of ketone body utilization characterized by intermittent ketoacidosis crises. This study reports the first Iranian patient with SCOT deficiency who presented with seizure and hypotonia at birth. Accordingly, she was consequently re-hospitalized due to hypotonia and respiratory distress. Laboratory tests revealed hyperammonemia, ketonuria, and metabolic acidosis. Besides, the plasma glucose level was normal without any other abnormality. Despite treatment with high-dose bicarbonate, severe acidosis persisted. Poor response to treatment raised a significant diagnostic challenge among specialists until genetic investigation identified a homozygous nonsense mutation (c.79G>T; p.Gly27*) in the OXCT1 gene (NM_000436), causing SCOT deficiency. Genetic studies help clinicians achieve a definite diagnosis of such metabolic disorders. In this case, the accurate and early diagnosis of SCOT deficiency opened new therapeutic possibilities, including frequent carbohydrate-rich meals and low fat and protein diet. Moreover, our findings expand the mutational and clinical spectrum of SCOT deficiency.



 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


