Ajay N. Jain, Alexander C. Brueckner, Christine Jorge, Ann E. Cleves, Purnima Khandelwal, Janet Caceres Cortes, Luciano Mueller
{"title":"复合肽大环优化:结合核磁共振约束和构象分析指导基于结构和配体的设计","authors":"Ajay N. Jain, Alexander C. Brueckner, Christine Jorge, Ann E. Cleves, Purnima Khandelwal, Janet Caceres Cortes, Luciano Mueller","doi":"10.1007/s10822-023-00524-2","DOIUrl":null,"url":null,"abstract":"<div><p>Systematic optimization of large macrocyclic peptide ligands is a serious challenge. Here, we describe an approach for lead-optimization using the PD-1/PD-L1 system as a retrospective example of moving from initial lead compound to clinical candidate. We show how conformational restraints can be derived by exploiting NMR data to identify low-energy solution ensembles of a lead compound. Such restraints can be used to focus conformational search for analogs in order to accurately predict bound ligand poses through molecular docking and thereby estimate ligand strain and protein-ligand intermolecular binding energy. We also describe an analogous ligand-based approach that employs molecular similarity optimization to predict bound poses. Both approaches are shown to be effective for prioritizing lead-compound analogs. Surprisingly, relatively small ligand modifications, which may have minimal effects on predicted bound pose or intermolecular interactions, often lead to large changes in estimated strain that have dominating effects on overall binding energy estimates. Effective macrocyclic conformational search is crucial, whether in the context of NMR-based restraints, X-ray ligand refinement, partial torsional restraint for docking/ligand-similarity calculations or agnostic search for nominal global minima. Lead optimization for peptidic macrocycles can be made more productive using a multi-disciplinary approach that combines biophysical data with practical and efficient computational methods.</p></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"37 11","pages":"519 - 535"},"PeriodicalIF":3.0000,"publicationDate":"2023-08-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s10822-023-00524-2.pdf","citationCount":"0","resultStr":"{\"title\":\"Complex peptide macrocycle optimization: combining NMR restraints with conformational analysis to guide structure-based and ligand-based design\",\"authors\":\"Ajay N. Jain, Alexander C. Brueckner, Christine Jorge, Ann E. Cleves, Purnima Khandelwal, Janet Caceres Cortes, Luciano Mueller\",\"doi\":\"10.1007/s10822-023-00524-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Systematic optimization of large macrocyclic peptide ligands is a serious challenge. Here, we describe an approach for lead-optimization using the PD-1/PD-L1 system as a retrospective example of moving from initial lead compound to clinical candidate. We show how conformational restraints can be derived by exploiting NMR data to identify low-energy solution ensembles of a lead compound. Such restraints can be used to focus conformational search for analogs in order to accurately predict bound ligand poses through molecular docking and thereby estimate ligand strain and protein-ligand intermolecular binding energy. We also describe an analogous ligand-based approach that employs molecular similarity optimization to predict bound poses. Both approaches are shown to be effective for prioritizing lead-compound analogs. Surprisingly, relatively small ligand modifications, which may have minimal effects on predicted bound pose or intermolecular interactions, often lead to large changes in estimated strain that have dominating effects on overall binding energy estimates. Effective macrocyclic conformational search is crucial, whether in the context of NMR-based restraints, X-ray ligand refinement, partial torsional restraint for docking/ligand-similarity calculations or agnostic search for nominal global minima. Lead optimization for peptidic macrocycles can be made more productive using a multi-disciplinary approach that combines biophysical data with practical and efficient computational methods.</p></div>\",\"PeriodicalId\":621,\"journal\":{\"name\":\"Journal of Computer-Aided Molecular Design\",\"volume\":\"37 11\",\"pages\":\"519 - 535\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2023-08-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://link.springer.com/content/pdf/10.1007/s10822-023-00524-2.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computer-Aided Molecular Design\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10822-023-00524-2\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-023-00524-2","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
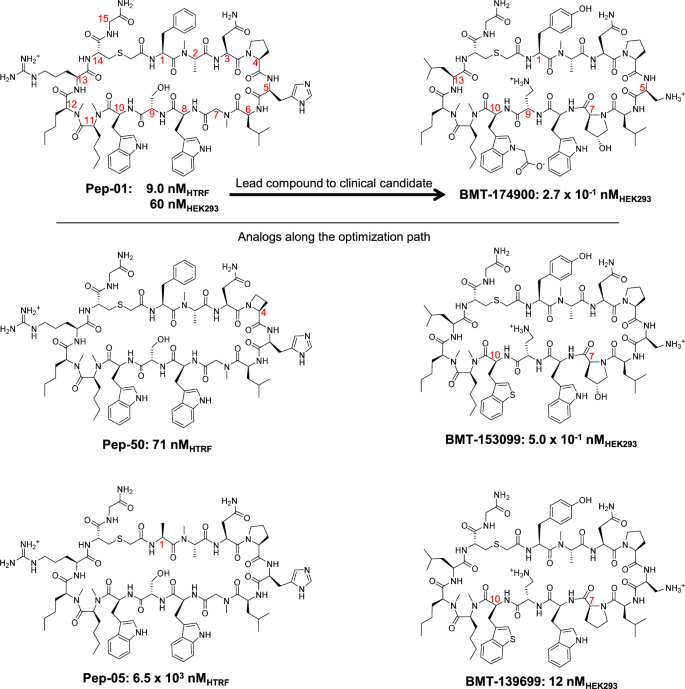
Complex peptide macrocycle optimization: combining NMR restraints with conformational analysis to guide structure-based and ligand-based design
Systematic optimization of large macrocyclic peptide ligands is a serious challenge. Here, we describe an approach for lead-optimization using the PD-1/PD-L1 system as a retrospective example of moving from initial lead compound to clinical candidate. We show how conformational restraints can be derived by exploiting NMR data to identify low-energy solution ensembles of a lead compound. Such restraints can be used to focus conformational search for analogs in order to accurately predict bound ligand poses through molecular docking and thereby estimate ligand strain and protein-ligand intermolecular binding energy. We also describe an analogous ligand-based approach that employs molecular similarity optimization to predict bound poses. Both approaches are shown to be effective for prioritizing lead-compound analogs. Surprisingly, relatively small ligand modifications, which may have minimal effects on predicted bound pose or intermolecular interactions, often lead to large changes in estimated strain that have dominating effects on overall binding energy estimates. Effective macrocyclic conformational search is crucial, whether in the context of NMR-based restraints, X-ray ligand refinement, partial torsional restraint for docking/ligand-similarity calculations or agnostic search for nominal global minima. Lead optimization for peptidic macrocycles can be made more productive using a multi-disciplinary approach that combines biophysical data with practical and efficient computational methods.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


