{"title":"adi - qsar:基于化合物生物活性差异的机器学习模型","authors":"Gyoung Jin Park, Nam Sook Kang","doi":"10.1007/s10822-023-00517-1","DOIUrl":null,"url":null,"abstract":"<div><p>Drug candidates identified by the pharmaceutical industry typically have unique structural characteristics to ensure they interact strongly and specifically with their biological targets. Identifying these characteristics is a key challenge for developing new drugs, and quantitative structure-activity relationship (QSAR) analysis has generally been used to perform this task. QSAR models with good predictive power improve the cost and time efficiencies invested in compound development. Generating these good models depends on how well differences between “active” and “inactive” compound groups can be conveyed to the model to be learned. Efforts to solve this difference issue have been made, including generating a “molecular descriptor” that compressively expresses the structural characteristics of compounds. From the same perspective, we succeeded in developing the Activity Differences-Quantitative Structure-Activity Relationship (ADis-QSAR) model by generating molecular descriptors that more explicitly convey features of the group through a pair system that performs direct connections between active and inactive groups. We used popular machine learning algorithms, such as Support Vector Machine, Random Forest, XGBoost and Multi-Layer Perceptron for model learning and evaluated the model using scores such as accuracy, area under curve, precision and specificity. The results showed that the Support Vector Machine performed better than the others. Notably, the ADis-QSAR model showed significant improvements in meaningful scores such as precision and specificity compared to the baseline model, even in datasets with dissimilar chemical spaces. This model reduces the risk of selecting false positive compounds, improving the efficiency of drug development.</p></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"37 9","pages":"435 - 451"},"PeriodicalIF":3.0000,"publicationDate":"2023-06-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"ADis-QSAR: a machine learning model based on biological activity differences of compounds\",\"authors\":\"Gyoung Jin Park, Nam Sook Kang\",\"doi\":\"10.1007/s10822-023-00517-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Drug candidates identified by the pharmaceutical industry typically have unique structural characteristics to ensure they interact strongly and specifically with their biological targets. Identifying these characteristics is a key challenge for developing new drugs, and quantitative structure-activity relationship (QSAR) analysis has generally been used to perform this task. QSAR models with good predictive power improve the cost and time efficiencies invested in compound development. Generating these good models depends on how well differences between “active” and “inactive” compound groups can be conveyed to the model to be learned. Efforts to solve this difference issue have been made, including generating a “molecular descriptor” that compressively expresses the structural characteristics of compounds. From the same perspective, we succeeded in developing the Activity Differences-Quantitative Structure-Activity Relationship (ADis-QSAR) model by generating molecular descriptors that more explicitly convey features of the group through a pair system that performs direct connections between active and inactive groups. We used popular machine learning algorithms, such as Support Vector Machine, Random Forest, XGBoost and Multi-Layer Perceptron for model learning and evaluated the model using scores such as accuracy, area under curve, precision and specificity. The results showed that the Support Vector Machine performed better than the others. Notably, the ADis-QSAR model showed significant improvements in meaningful scores such as precision and specificity compared to the baseline model, even in datasets with dissimilar chemical spaces. This model reduces the risk of selecting false positive compounds, improving the efficiency of drug development.</p></div>\",\"PeriodicalId\":621,\"journal\":{\"name\":\"Journal of Computer-Aided Molecular Design\",\"volume\":\"37 9\",\"pages\":\"435 - 451\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2023-06-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computer-Aided Molecular Design\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10822-023-00517-1\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-023-00517-1","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
ADis-QSAR: a machine learning model based on biological activity differences of compounds
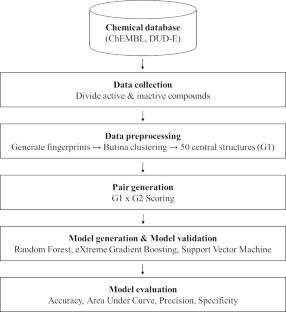
Drug candidates identified by the pharmaceutical industry typically have unique structural characteristics to ensure they interact strongly and specifically with their biological targets. Identifying these characteristics is a key challenge for developing new drugs, and quantitative structure-activity relationship (QSAR) analysis has generally been used to perform this task. QSAR models with good predictive power improve the cost and time efficiencies invested in compound development. Generating these good models depends on how well differences between “active” and “inactive” compound groups can be conveyed to the model to be learned. Efforts to solve this difference issue have been made, including generating a “molecular descriptor” that compressively expresses the structural characteristics of compounds. From the same perspective, we succeeded in developing the Activity Differences-Quantitative Structure-Activity Relationship (ADis-QSAR) model by generating molecular descriptors that more explicitly convey features of the group through a pair system that performs direct connections between active and inactive groups. We used popular machine learning algorithms, such as Support Vector Machine, Random Forest, XGBoost and Multi-Layer Perceptron for model learning and evaluated the model using scores such as accuracy, area under curve, precision and specificity. The results showed that the Support Vector Machine performed better than the others. Notably, the ADis-QSAR model showed significant improvements in meaningful scores such as precision and specificity compared to the baseline model, even in datasets with dissimilar chemical spaces. This model reduces the risk of selecting false positive compounds, improving the efficiency of drug development.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


