Wemenes José Lima Silva, Renato Ferreira de Freitas
{"title":"评估对接、FEP和MM/GBSA方法对一系列KLK6抑制剂的性能","authors":"Wemenes José Lima Silva, Renato Ferreira de Freitas","doi":"10.1007/s10822-023-00515-3","DOIUrl":null,"url":null,"abstract":"<div><p>Kallikrein 6 (KLK6) is an attractive drug target for the treatment of neurological diseases and for various cancers. Herein, we explore the accuracy and efficiency of different computational methods and protocols to predict the free energy of binding (ΔG<sub>bind</sub>) for a series of 49 inhibitors of KLK6. We found that the performance of the methods varied strongly with the tested system. For only one of the three KLK6 datasets, the docking scores obtained with rDock were in good agreement (R<sup>2</sup> ≥ 0.5) with experimental values of ΔG<sub>bind</sub>. A similar result was obtained with MM/GBSA (using the ff14SB force field) calculations based on single minimized structures. Improved binding affinity predictions were obtained with the free energy perturbation (FEP) method, with an overall MUE and RMSE of 0.53 and 0.68 kcal/mol, respectively. Furthermore, in a simulation of a real-world drug discovery project, FEP was able to rank the most potent compounds at the top of the list. These results indicate that FEP can be a promising tool for the structure-based optimization of KLK6 inhibitors.</p></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"37 9","pages":"407 - 418"},"PeriodicalIF":3.0000,"publicationDate":"2023-06-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s10822-023-00515-3.pdf","citationCount":"0","resultStr":"{\"title\":\"Assessing the performance of docking, FEP, and MM/GBSA methods on a series of KLK6 inhibitors\",\"authors\":\"Wemenes José Lima Silva, Renato Ferreira de Freitas\",\"doi\":\"10.1007/s10822-023-00515-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Kallikrein 6 (KLK6) is an attractive drug target for the treatment of neurological diseases and for various cancers. Herein, we explore the accuracy and efficiency of different computational methods and protocols to predict the free energy of binding (ΔG<sub>bind</sub>) for a series of 49 inhibitors of KLK6. We found that the performance of the methods varied strongly with the tested system. For only one of the three KLK6 datasets, the docking scores obtained with rDock were in good agreement (R<sup>2</sup> ≥ 0.5) with experimental values of ΔG<sub>bind</sub>. A similar result was obtained with MM/GBSA (using the ff14SB force field) calculations based on single minimized structures. Improved binding affinity predictions were obtained with the free energy perturbation (FEP) method, with an overall MUE and RMSE of 0.53 and 0.68 kcal/mol, respectively. Furthermore, in a simulation of a real-world drug discovery project, FEP was able to rank the most potent compounds at the top of the list. These results indicate that FEP can be a promising tool for the structure-based optimization of KLK6 inhibitors.</p></div>\",\"PeriodicalId\":621,\"journal\":{\"name\":\"Journal of Computer-Aided Molecular Design\",\"volume\":\"37 9\",\"pages\":\"407 - 418\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2023-06-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://link.springer.com/content/pdf/10.1007/s10822-023-00515-3.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computer-Aided Molecular Design\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10822-023-00515-3\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-023-00515-3","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Assessing the performance of docking, FEP, and MM/GBSA methods on a series of KLK6 inhibitors
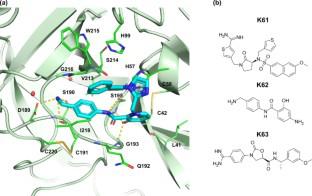
Kallikrein 6 (KLK6) is an attractive drug target for the treatment of neurological diseases and for various cancers. Herein, we explore the accuracy and efficiency of different computational methods and protocols to predict the free energy of binding (ΔGbind) for a series of 49 inhibitors of KLK6. We found that the performance of the methods varied strongly with the tested system. For only one of the three KLK6 datasets, the docking scores obtained with rDock were in good agreement (R2 ≥ 0.5) with experimental values of ΔGbind. A similar result was obtained with MM/GBSA (using the ff14SB force field) calculations based on single minimized structures. Improved binding affinity predictions were obtained with the free energy perturbation (FEP) method, with an overall MUE and RMSE of 0.53 and 0.68 kcal/mol, respectively. Furthermore, in a simulation of a real-world drug discovery project, FEP was able to rank the most potent compounds at the top of the list. These results indicate that FEP can be a promising tool for the structure-based optimization of KLK6 inhibitors.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


