Elizabeth G. Gibson, Ben Bax*, Pan F. Chan, Neil Osheroff*
{"title":"抗菌药物Gepotidacin抗金黄色葡萄球菌旋转酶作用的机制和结构基础","authors":"Elizabeth G. Gibson, Ben Bax*, Pan F. Chan, Neil Osheroff*","doi":"10.1021/acsinfecdis.8b00315","DOIUrl":null,"url":null,"abstract":"<p >Gepotidacin is a first-in-class triazaacenaphthylene novel bacterial topoisomerase inhibitor (NBTI). The compound has successfully completed phase II trials for the treatment of acute bacterial skin/skin structure infections and for the treatment of uncomplicated urogenital gonorrhea. It also displays robust <i>in vitro</i> activity against a range of wild-type and fluoroquinolone-resistant bacteria. Due to the clinical promise of gepotidacin, a detailed understanding of its interactions with its antibacterial targets is essential. Thus, we characterized the mechanism of action of gepotidacin against <i>Staphylococcus aureus</i> gyrase. Gepotidacin was a potent inhibitor of gyrase-catalyzed DNA supercoiling (IC<sub>50</sub> ≈ 0.047 μM) and relaxation of positively supercoiled substrates (IC<sub>50</sub> ≈ 0.6 μM). Unlike fluoroquinolones, which induce primarily double-stranded DNA breaks, gepotidacin induced high levels of gyrase-mediated single-stranded breaks. No double-stranded breaks were observed even at high gepotidacin concentration, long cleavage times, or in the presence of ATP. Moreover, gepotidacin suppressed the formation of double-stranded breaks. Gepotidacin formed gyrase–DNA cleavage complexes that were stable for >4 h. <i>In vitro</i> competition suggests that gyrase binding by gepotidacin and fluoroquinolones are mutually exclusive. Finally, we determined crystal structures of gepotidacin with the <i>S. aureus</i> gyrase core fusion truncate with nicked (2.31 ? resolution) or intact (uncleaved) DNA (2.37 ? resolution). In both cases, a single gepotidacin molecule was bound midway between the two scissile DNA bonds and in a pocket between the two GyrA subunits. A comparison of the two structures demonstrates conformational flexibility within the central linker of gepotidacin, which may contribute to the activity of the compound.</p>","PeriodicalId":17,"journal":{"name":"ACS Infectious Diseases","volume":"5 4","pages":"570–581"},"PeriodicalIF":3.8000,"publicationDate":"2019-02-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1021/acsinfecdis.8b00315","citationCount":"92","resultStr":"{\"title\":\"Mechanistic and Structural Basis for the Actions of the Antibacterial Gepotidacin against Staphylococcus aureus Gyrase\",\"authors\":\"Elizabeth G. Gibson, Ben Bax*, Pan F. Chan, Neil Osheroff*\",\"doi\":\"10.1021/acsinfecdis.8b00315\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Gepotidacin is a first-in-class triazaacenaphthylene novel bacterial topoisomerase inhibitor (NBTI). The compound has successfully completed phase II trials for the treatment of acute bacterial skin/skin structure infections and for the treatment of uncomplicated urogenital gonorrhea. It also displays robust <i>in vitro</i> activity against a range of wild-type and fluoroquinolone-resistant bacteria. Due to the clinical promise of gepotidacin, a detailed understanding of its interactions with its antibacterial targets is essential. Thus, we characterized the mechanism of action of gepotidacin against <i>Staphylococcus aureus</i> gyrase. Gepotidacin was a potent inhibitor of gyrase-catalyzed DNA supercoiling (IC<sub>50</sub> ≈ 0.047 μM) and relaxation of positively supercoiled substrates (IC<sub>50</sub> ≈ 0.6 μM). Unlike fluoroquinolones, which induce primarily double-stranded DNA breaks, gepotidacin induced high levels of gyrase-mediated single-stranded breaks. No double-stranded breaks were observed even at high gepotidacin concentration, long cleavage times, or in the presence of ATP. Moreover, gepotidacin suppressed the formation of double-stranded breaks. Gepotidacin formed gyrase–DNA cleavage complexes that were stable for >4 h. <i>In vitro</i> competition suggests that gyrase binding by gepotidacin and fluoroquinolones are mutually exclusive. Finally, we determined crystal structures of gepotidacin with the <i>S. aureus</i> gyrase core fusion truncate with nicked (2.31 ? resolution) or intact (uncleaved) DNA (2.37 ? resolution). In both cases, a single gepotidacin molecule was bound midway between the two scissile DNA bonds and in a pocket between the two GyrA subunits. A comparison of the two structures demonstrates conformational flexibility within the central linker of gepotidacin, which may contribute to the activity of the compound.</p>\",\"PeriodicalId\":17,\"journal\":{\"name\":\"ACS Infectious Diseases\",\"volume\":\"5 4\",\"pages\":\"570–581\"},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2019-02-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1021/acsinfecdis.8b00315\",\"citationCount\":\"92\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Infectious Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acsinfecdis.8b00315\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Infectious Diseases","FirstCategoryId":"3","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acsinfecdis.8b00315","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Mechanistic and Structural Basis for the Actions of the Antibacterial Gepotidacin against Staphylococcus aureus Gyrase
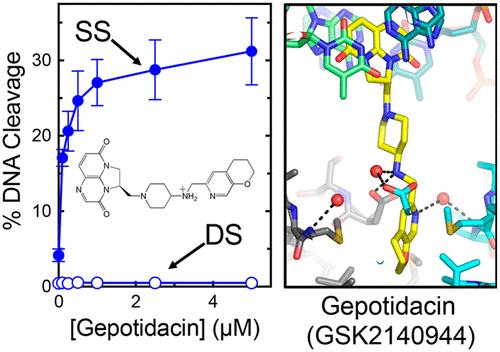
Gepotidacin is a first-in-class triazaacenaphthylene novel bacterial topoisomerase inhibitor (NBTI). The compound has successfully completed phase II trials for the treatment of acute bacterial skin/skin structure infections and for the treatment of uncomplicated urogenital gonorrhea. It also displays robust in vitro activity against a range of wild-type and fluoroquinolone-resistant bacteria. Due to the clinical promise of gepotidacin, a detailed understanding of its interactions with its antibacterial targets is essential. Thus, we characterized the mechanism of action of gepotidacin against Staphylococcus aureus gyrase. Gepotidacin was a potent inhibitor of gyrase-catalyzed DNA supercoiling (IC50 ≈ 0.047 μM) and relaxation of positively supercoiled substrates (IC50 ≈ 0.6 μM). Unlike fluoroquinolones, which induce primarily double-stranded DNA breaks, gepotidacin induced high levels of gyrase-mediated single-stranded breaks. No double-stranded breaks were observed even at high gepotidacin concentration, long cleavage times, or in the presence of ATP. Moreover, gepotidacin suppressed the formation of double-stranded breaks. Gepotidacin formed gyrase–DNA cleavage complexes that were stable for >4 h. In vitro competition suggests that gyrase binding by gepotidacin and fluoroquinolones are mutually exclusive. Finally, we determined crystal structures of gepotidacin with the S. aureus gyrase core fusion truncate with nicked (2.31 ? resolution) or intact (uncleaved) DNA (2.37 ? resolution). In both cases, a single gepotidacin molecule was bound midway between the two scissile DNA bonds and in a pocket between the two GyrA subunits. A comparison of the two structures demonstrates conformational flexibility within the central linker of gepotidacin, which may contribute to the activity of the compound.
期刊介绍:
ACS Infectious Diseases will be the first journal to highlight chemistry and its role in this multidisciplinary and collaborative research area. The journal will cover a diverse array of topics including, but not limited to:
* Discovery and development of new antimicrobial agents — identified through target- or phenotypic-based approaches as well as compounds that induce synergy with antimicrobials.
* Characterization and validation of drug target or pathways — use of single target and genome-wide knockdown and knockouts, biochemical studies, structural biology, new technologies to facilitate characterization and prioritization of potential drug targets.
* Mechanism of drug resistance — fundamental research that advances our understanding of resistance; strategies to prevent resistance.
* Mechanisms of action — use of genetic, metabolomic, and activity- and affinity-based protein profiling to elucidate the mechanism of action of clinical and experimental antimicrobial agents.
* Host-pathogen interactions — tools for studying host-pathogen interactions, cellular biochemistry of hosts and pathogens, and molecular interactions of pathogens with host microbiota.
* Small molecule vaccine adjuvants for infectious disease.
* Viral and bacterial biochemistry and molecular biology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


