Roberto Paciotti, Cecilia Coletti, Alessandro Marrone, Nazzareno Re
{"title":"配体-受体结合能的FMO2分析:比斯卡宾-金(I)/DNA g -四重体案例研究","authors":"Roberto Paciotti, Cecilia Coletti, Alessandro Marrone, Nazzareno Re","doi":"10.1007/s10822-022-00484-z","DOIUrl":null,"url":null,"abstract":"<div><p>In this work, the ab initio fragment molecular orbital (FMO) method was applied to calculate and analyze the binding energy of two biscarbene-Au(I) derivatives, [Au(9-methylcaffein-8-ylidene)<sub>2</sub>]<sup>+</sup> and [Au(1,3-dimethylbenzimidazol-2-ylidene)<sub>2</sub>]<sup>+</sup>, to the DNA G-Quadruplex structure. The FMO2 binding energy considers the ligand-receptor complex as well as the isolated forms of energy-minimum state of ligand and receptor, providing a better description of ligand-receptor affinity compared with simple pair interaction energies (PIE). Our results highlight important features of the binding process of biscarbene-Au(I) derivatives to DNA G-Quadruplex, indicating that the total deformation-polarization energy and desolvation penalty of the ligands are the main terms destabilizing the binding. The pair interaction energy decomposition analysis (PIEDA) between ligand and nucleobases suggest that the main interaction terms are electrostatic and charge-transfer energies supporting the hypothesis that Au(I) ion can be involved in π-cation interactions further stabilizing the ligand-receptor complex. Moreover, the presence of polar groups on the carbene ring, as C = O, can improve the charge-transfer interaction with K<sup>+</sup> ion. These findings can be employed to design new powerful biscarbene-Au(I) DNA-G quadruplex binders as promising anticancer drugs. The procedure described in this work can be applied to investigate any ligand-receptor system and is particularly useful when the binding process is strongly characterized by polarization, charge-transfer and dispersion interactions, properly evaluated by ab initio methods.</p></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"36 12","pages":"851 - 866"},"PeriodicalIF":3.0000,"publicationDate":"2022-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s10822-022-00484-z.pdf","citationCount":"2","resultStr":"{\"title\":\"The FMO2 analysis of the ligand-receptor binding energy: the Biscarbene-Gold(I)/DNA G-Quadruplex case study\",\"authors\":\"Roberto Paciotti, Cecilia Coletti, Alessandro Marrone, Nazzareno Re\",\"doi\":\"10.1007/s10822-022-00484-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>In this work, the ab initio fragment molecular orbital (FMO) method was applied to calculate and analyze the binding energy of two biscarbene-Au(I) derivatives, [Au(9-methylcaffein-8-ylidene)<sub>2</sub>]<sup>+</sup> and [Au(1,3-dimethylbenzimidazol-2-ylidene)<sub>2</sub>]<sup>+</sup>, to the DNA G-Quadruplex structure. The FMO2 binding energy considers the ligand-receptor complex as well as the isolated forms of energy-minimum state of ligand and receptor, providing a better description of ligand-receptor affinity compared with simple pair interaction energies (PIE). Our results highlight important features of the binding process of biscarbene-Au(I) derivatives to DNA G-Quadruplex, indicating that the total deformation-polarization energy and desolvation penalty of the ligands are the main terms destabilizing the binding. The pair interaction energy decomposition analysis (PIEDA) between ligand and nucleobases suggest that the main interaction terms are electrostatic and charge-transfer energies supporting the hypothesis that Au(I) ion can be involved in π-cation interactions further stabilizing the ligand-receptor complex. Moreover, the presence of polar groups on the carbene ring, as C = O, can improve the charge-transfer interaction with K<sup>+</sup> ion. These findings can be employed to design new powerful biscarbene-Au(I) DNA-G quadruplex binders as promising anticancer drugs. The procedure described in this work can be applied to investigate any ligand-receptor system and is particularly useful when the binding process is strongly characterized by polarization, charge-transfer and dispersion interactions, properly evaluated by ab initio methods.</p></div>\",\"PeriodicalId\":621,\"journal\":{\"name\":\"Journal of Computer-Aided Molecular Design\",\"volume\":\"36 12\",\"pages\":\"851 - 866\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2022-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://link.springer.com/content/pdf/10.1007/s10822-022-00484-z.pdf\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computer-Aided Molecular Design\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10822-022-00484-z\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-022-00484-z","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 2
摘要
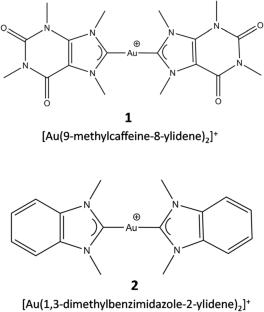
本文采用从头算片段分子轨道(FMO)方法,计算并分析了两种双卡宾-Au(I)衍生物[Au(9-甲基咖啡因-8-酰基)2]+和[Au(1,3-二甲基苯并咪唑-2-酰基)2]+对DNA g -四重体结构的结合能。FMO2结合能考虑了配体-受体复合物以及配体和受体能量最低状态的孤立形式,与简单对相互作用能(PIE)相比,能更好地描述配体-受体的亲和力。我们的研究结果突出了比斯卡宾-金(I)衍生物与DNA g -四重体结合过程的重要特征,表明配体的总变形极化能和脱溶惩罚是破坏结合的主要因素。配体与核碱基之间的对相互作用能分解分析(PIEDA)表明,主要相互作用项是静电能和电荷转移能,支持Au(I)离子可以参与π-阳离子相互作用的假设,进一步稳定配体-受体复合物。此外,碳环上存在极性基团,如C = O,可以改善与K+离子的电荷转移相互作用。这些发现可用于设计新的强效双卡宾- au (I) DNA-G四重体结合物,作为有前景的抗癌药物。这项工作中描述的程序可以应用于研究任何配体-受体系统,当结合过程具有极化、电荷转移和色散相互作用的强烈特征时,特别有用,可以用从头算方法进行适当的评估。
The FMO2 analysis of the ligand-receptor binding energy: the Biscarbene-Gold(I)/DNA G-Quadruplex case study
In this work, the ab initio fragment molecular orbital (FMO) method was applied to calculate and analyze the binding energy of two biscarbene-Au(I) derivatives, [Au(9-methylcaffein-8-ylidene)2]+ and [Au(1,3-dimethylbenzimidazol-2-ylidene)2]+, to the DNA G-Quadruplex structure. The FMO2 binding energy considers the ligand-receptor complex as well as the isolated forms of energy-minimum state of ligand and receptor, providing a better description of ligand-receptor affinity compared with simple pair interaction energies (PIE). Our results highlight important features of the binding process of biscarbene-Au(I) derivatives to DNA G-Quadruplex, indicating that the total deformation-polarization energy and desolvation penalty of the ligands are the main terms destabilizing the binding. The pair interaction energy decomposition analysis (PIEDA) between ligand and nucleobases suggest that the main interaction terms are electrostatic and charge-transfer energies supporting the hypothesis that Au(I) ion can be involved in π-cation interactions further stabilizing the ligand-receptor complex. Moreover, the presence of polar groups on the carbene ring, as C = O, can improve the charge-transfer interaction with K+ ion. These findings can be employed to design new powerful biscarbene-Au(I) DNA-G quadruplex binders as promising anticancer drugs. The procedure described in this work can be applied to investigate any ligand-receptor system and is particularly useful when the binding process is strongly characterized by polarization, charge-transfer and dispersion interactions, properly evaluated by ab initio methods.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


