Ernesto Pavoni, Francesca Sciandra, Giorgio Tasca, Roberta Tittarelli, Manuela Bozzi, Bruno Giardina, Enzo Ricci, Andrea Brancaccio
{"title":"糖营养不良亚基的免疫学分析:非先天性营养不良患者小队列的经验教训。","authors":"Ernesto Pavoni, Francesca Sciandra, Giorgio Tasca, Roberta Tittarelli, Manuela Bozzi, Bruno Giardina, Enzo Ricci, Andrea Brancaccio","doi":"10.2174/1874205X01105010068","DOIUrl":null,"url":null,"abstract":"<p><p>The dystroglycan (DG) expression pattern can be altered in severe muscular dystrophies. In fact, some congenital muscular dystrophies (CMDs) and limb-girdle muscular dystrophies (LGMDs) are caused by point mutations identified in six glycosyltransferase genes which are likely to target different steps along the posttranslational \"O-glycosylation route\" leading to a fully decorated and functional α-DG subunit. Indeed, hypoglycosylation of α-DG is thought to represent a major pathological event, in that it could reduce the DG's ability to bind the basement membrane components, thus leading to sarcolemmal instability and necrosis. In order to set up an efficient standard immunological protocol, taking advantage of a wide panel of antibodies, we have analyzed the two DG subunits in a small cohort of adult dystrophic patients, whom an extensive medical examination had already clinically classified as affected by LGMD (5), Miyoshi (1) or distal (1) myopathy. Immunofluorescence analysis of skeletal muscle tissue sections revealed a proper sarcolemmal localization of the DG subunits in all the patients analyzed. However, Western blot analysis of lectin enriched skeletal muscle samples revealed an abnormal glycosylation of α-DG in two patients. Our work reinforces the notion that a careful immunological and biochemical analysis of the two DG subunits should be always considered as a prerequisite for the identification of new putative cases of dystroglycanopathy.</p>","PeriodicalId":39123,"journal":{"name":"Open Neurology Journal","volume":" ","pages":"68-74"},"PeriodicalIF":0.0000,"publicationDate":"2011-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/8e/b1/TONEUJ-5-68.PMC3204415.pdf","citationCount":"0","resultStr":"{\"title\":\"An immunological analysis of dystroglycan subunits: lessons learned from a small cohort of non-congenital dystrophic patients.\",\"authors\":\"Ernesto Pavoni, Francesca Sciandra, Giorgio Tasca, Roberta Tittarelli, Manuela Bozzi, Bruno Giardina, Enzo Ricci, Andrea Brancaccio\",\"doi\":\"10.2174/1874205X01105010068\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The dystroglycan (DG) expression pattern can be altered in severe muscular dystrophies. In fact, some congenital muscular dystrophies (CMDs) and limb-girdle muscular dystrophies (LGMDs) are caused by point mutations identified in six glycosyltransferase genes which are likely to target different steps along the posttranslational \\\"O-glycosylation route\\\" leading to a fully decorated and functional α-DG subunit. Indeed, hypoglycosylation of α-DG is thought to represent a major pathological event, in that it could reduce the DG's ability to bind the basement membrane components, thus leading to sarcolemmal instability and necrosis. In order to set up an efficient standard immunological protocol, taking advantage of a wide panel of antibodies, we have analyzed the two DG subunits in a small cohort of adult dystrophic patients, whom an extensive medical examination had already clinically classified as affected by LGMD (5), Miyoshi (1) or distal (1) myopathy. Immunofluorescence analysis of skeletal muscle tissue sections revealed a proper sarcolemmal localization of the DG subunits in all the patients analyzed. However, Western blot analysis of lectin enriched skeletal muscle samples revealed an abnormal glycosylation of α-DG in two patients. Our work reinforces the notion that a careful immunological and biochemical analysis of the two DG subunits should be always considered as a prerequisite for the identification of new putative cases of dystroglycanopathy.</p>\",\"PeriodicalId\":39123,\"journal\":{\"name\":\"Open Neurology Journal\",\"volume\":\" \",\"pages\":\"68-74\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2011-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/8e/b1/TONEUJ-5-68.PMC3204415.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Open Neurology Journal\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.2174/1874205X01105010068\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2011/10/20 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Open Neurology Journal","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2174/1874205X01105010068","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2011/10/20 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
An immunological analysis of dystroglycan subunits: lessons learned from a small cohort of non-congenital dystrophic patients.
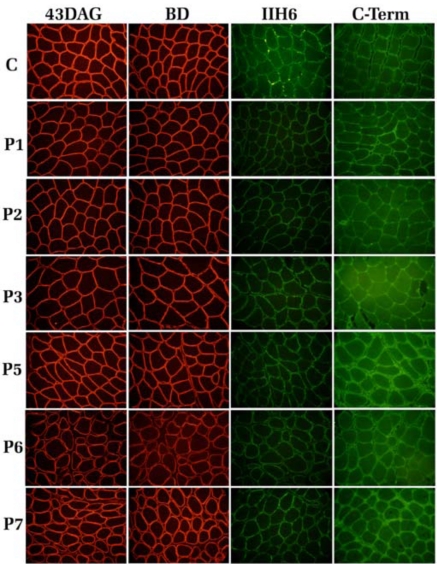
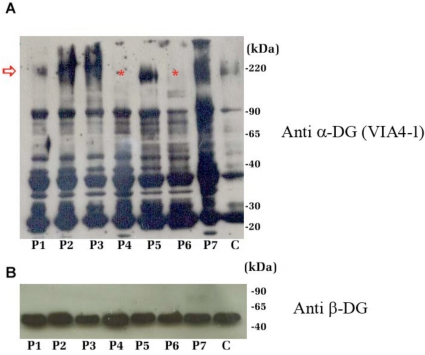
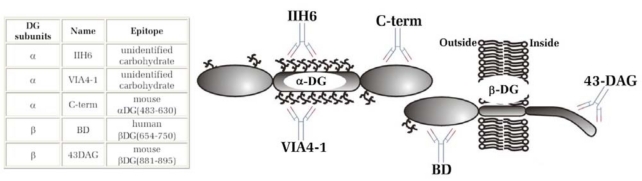
The dystroglycan (DG) expression pattern can be altered in severe muscular dystrophies. In fact, some congenital muscular dystrophies (CMDs) and limb-girdle muscular dystrophies (LGMDs) are caused by point mutations identified in six glycosyltransferase genes which are likely to target different steps along the posttranslational "O-glycosylation route" leading to a fully decorated and functional α-DG subunit. Indeed, hypoglycosylation of α-DG is thought to represent a major pathological event, in that it could reduce the DG's ability to bind the basement membrane components, thus leading to sarcolemmal instability and necrosis. In order to set up an efficient standard immunological protocol, taking advantage of a wide panel of antibodies, we have analyzed the two DG subunits in a small cohort of adult dystrophic patients, whom an extensive medical examination had already clinically classified as affected by LGMD (5), Miyoshi (1) or distal (1) myopathy. Immunofluorescence analysis of skeletal muscle tissue sections revealed a proper sarcolemmal localization of the DG subunits in all the patients analyzed. However, Western blot analysis of lectin enriched skeletal muscle samples revealed an abnormal glycosylation of α-DG in two patients. Our work reinforces the notion that a careful immunological and biochemical analysis of the two DG subunits should be always considered as a prerequisite for the identification of new putative cases of dystroglycanopathy.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


