{"title":"一个预训练的硫化物固体电解质深电位模型,覆盖范围广,精度高","authors":"Ruoyu Wang, Mingyu Guo, Yuxiang Gao, Xiaoxu Wang, Yuzhi Zhang, Bin Deng, Mengchao Shi, Linfeng Zhang, Zhicheng Zhong","doi":"10.1038/s41524-025-01764-6","DOIUrl":null,"url":null,"abstract":"<p>Solid electrolytes with fast ion transport are crucial for solid state lithium metal batteries. Chemical doping has been the most effective strategy for improving ion condictiviy, and atomistic simulation with machine-learning potentials helps optimize doping by predicting ion conductivity for various composition. Yet most existing machine-learning models are trained on narrow chemistry, requiring retraining for each new system, which wastes transferable knowledge and incurs significant cost. Here, we propose a pre-trained deep potential model purpose-built for sulfide solid electrolytes with attention mechanism, known as DPA-SSE. The training set includes 15 elements and consists of both equilibrium and extensive out-of-equilibrium configurations. DPA-SSE achieves a high energy resolution of less than 2 meV/atom for dynamical trajectories up to 1150 K, and reproduces experimental ion conductivity with remarkable accuracy. DPA-SSE generalizes well to complex electrolytes with mixes of cation and anion atoms, and enables highly efficient dynamical simulation via model distillation. DPA-SSE also serves as a platform for continuous learning and can be fine-tuned with minimal downstream data. These results demonstrate the possibility of a new pathway for the AI-driven development of solid electrolytes with exceptional performance.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"52 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2025-08-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A pre-trained deep potential model for sulfide solid electrolytes with broad coverage and high accuracy\",\"authors\":\"Ruoyu Wang, Mingyu Guo, Yuxiang Gao, Xiaoxu Wang, Yuzhi Zhang, Bin Deng, Mengchao Shi, Linfeng Zhang, Zhicheng Zhong\",\"doi\":\"10.1038/s41524-025-01764-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Solid electrolytes with fast ion transport are crucial for solid state lithium metal batteries. Chemical doping has been the most effective strategy for improving ion condictiviy, and atomistic simulation with machine-learning potentials helps optimize doping by predicting ion conductivity for various composition. Yet most existing machine-learning models are trained on narrow chemistry, requiring retraining for each new system, which wastes transferable knowledge and incurs significant cost. Here, we propose a pre-trained deep potential model purpose-built for sulfide solid electrolytes with attention mechanism, known as DPA-SSE. The training set includes 15 elements and consists of both equilibrium and extensive out-of-equilibrium configurations. DPA-SSE achieves a high energy resolution of less than 2 meV/atom for dynamical trajectories up to 1150 K, and reproduces experimental ion conductivity with remarkable accuracy. DPA-SSE generalizes well to complex electrolytes with mixes of cation and anion atoms, and enables highly efficient dynamical simulation via model distillation. DPA-SSE also serves as a platform for continuous learning and can be fine-tuned with minimal downstream data. These results demonstrate the possibility of a new pathway for the AI-driven development of solid electrolytes with exceptional performance.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"52 1\",\"pages\":\"\"},\"PeriodicalIF\":11.9000,\"publicationDate\":\"2025-08-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-025-01764-6\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-025-01764-6","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
A pre-trained deep potential model for sulfide solid electrolytes with broad coverage and high accuracy
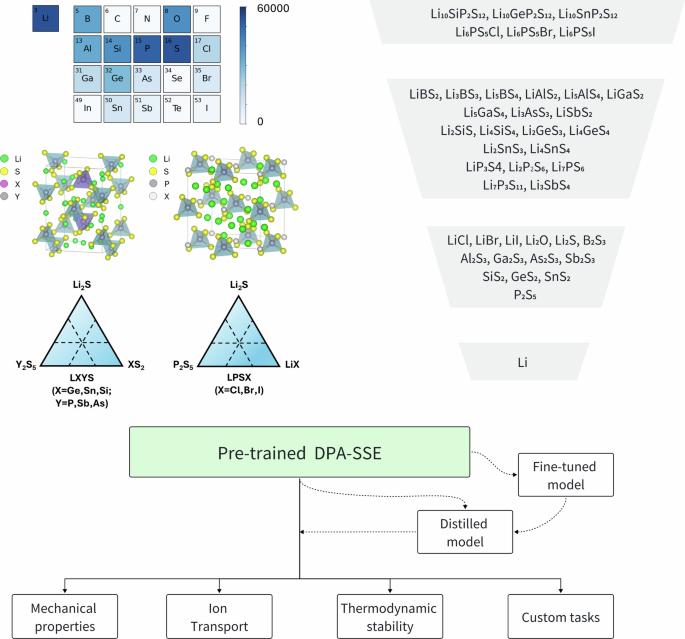
Solid electrolytes with fast ion transport are crucial for solid state lithium metal batteries. Chemical doping has been the most effective strategy for improving ion condictiviy, and atomistic simulation with machine-learning potentials helps optimize doping by predicting ion conductivity for various composition. Yet most existing machine-learning models are trained on narrow chemistry, requiring retraining for each new system, which wastes transferable knowledge and incurs significant cost. Here, we propose a pre-trained deep potential model purpose-built for sulfide solid electrolytes with attention mechanism, known as DPA-SSE. The training set includes 15 elements and consists of both equilibrium and extensive out-of-equilibrium configurations. DPA-SSE achieves a high energy resolution of less than 2 meV/atom for dynamical trajectories up to 1150 K, and reproduces experimental ion conductivity with remarkable accuracy. DPA-SSE generalizes well to complex electrolytes with mixes of cation and anion atoms, and enables highly efficient dynamical simulation via model distillation. DPA-SSE also serves as a platform for continuous learning and can be fine-tuned with minimal downstream data. These results demonstrate the possibility of a new pathway for the AI-driven development of solid electrolytes with exceptional performance.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


