Genming Lai, Ruiqi Zhang, Chi Fang, Juntao Zhao, Taowen Chen, Yunxing Zuo, Bo Xu, Jiaxin Zheng
{"title":"机器学习加速下锂金属阳极界面修饰机理探索","authors":"Genming Lai, Ruiqi Zhang, Chi Fang, Juntao Zhao, Taowen Chen, Yunxing Zuo, Bo Xu, Jiaxin Zheng","doi":"10.1038/s41524-025-01747-7","DOIUrl":null,"url":null,"abstract":"<p>Although the electrode-electrolyte interface is a crucial electrochemical region, the comprehensive understanding of interface reactions is limited by the time and space scales of experimental tools. Theoretical simulations with this delicate interface also remain one of the most significant challenges for atomistic modeling, particularly for the stable long-timescale simulation of the interface. Here we introduce a novel scheme, hybrid ab initio molecular dynamics combined with machine learning potential (HAML), to accelerate the modeling of electrode-electrolyte interface reactions. We demonstrate its effectiveness in modeling the interfaces of Li metal with both liquid and solid-state electrolytes, capturing critical processes over extended time scales. Furthermore, we reveal the role of interface reaction kinetics in interface regulation through HAML simulations, combined with the similarity analysis method. It is demonstrated that element (Se, F, O) doping in the Li<sub>6</sub>PS<sub>5</sub>Cl system is an effective strategy for enhancing interface reaction kinetics, facilitating the formation of a more stable interface protective layer faster at room temperature. Moreover, moderate structural instability can positively contribute to interface stabilization. HAML offers a promising approach for addressing the challenge of designing stable interfaces while reducing computational costs. This work provides valuable insights for advancing the understanding and optimization of interface behaviors in Li metal batteries.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"1 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2025-07-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Machine-learning-accelerated mechanistic exploration of interface modification in lithium metal anode\",\"authors\":\"Genming Lai, Ruiqi Zhang, Chi Fang, Juntao Zhao, Taowen Chen, Yunxing Zuo, Bo Xu, Jiaxin Zheng\",\"doi\":\"10.1038/s41524-025-01747-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Although the electrode-electrolyte interface is a crucial electrochemical region, the comprehensive understanding of interface reactions is limited by the time and space scales of experimental tools. Theoretical simulations with this delicate interface also remain one of the most significant challenges for atomistic modeling, particularly for the stable long-timescale simulation of the interface. Here we introduce a novel scheme, hybrid ab initio molecular dynamics combined with machine learning potential (HAML), to accelerate the modeling of electrode-electrolyte interface reactions. We demonstrate its effectiveness in modeling the interfaces of Li metal with both liquid and solid-state electrolytes, capturing critical processes over extended time scales. Furthermore, we reveal the role of interface reaction kinetics in interface regulation through HAML simulations, combined with the similarity analysis method. It is demonstrated that element (Se, F, O) doping in the Li<sub>6</sub>PS<sub>5</sub>Cl system is an effective strategy for enhancing interface reaction kinetics, facilitating the formation of a more stable interface protective layer faster at room temperature. Moreover, moderate structural instability can positively contribute to interface stabilization. HAML offers a promising approach for addressing the challenge of designing stable interfaces while reducing computational costs. This work provides valuable insights for advancing the understanding and optimization of interface behaviors in Li metal batteries.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"1 1\",\"pages\":\"\"},\"PeriodicalIF\":11.9000,\"publicationDate\":\"2025-07-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-025-01747-7\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-025-01747-7","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Machine-learning-accelerated mechanistic exploration of interface modification in lithium metal anode
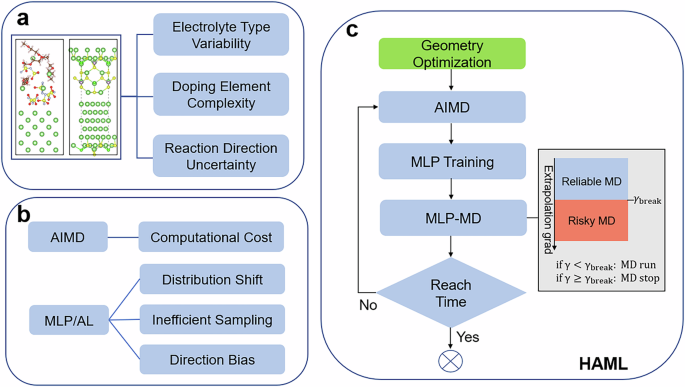
Although the electrode-electrolyte interface is a crucial electrochemical region, the comprehensive understanding of interface reactions is limited by the time and space scales of experimental tools. Theoretical simulations with this delicate interface also remain one of the most significant challenges for atomistic modeling, particularly for the stable long-timescale simulation of the interface. Here we introduce a novel scheme, hybrid ab initio molecular dynamics combined with machine learning potential (HAML), to accelerate the modeling of electrode-electrolyte interface reactions. We demonstrate its effectiveness in modeling the interfaces of Li metal with both liquid and solid-state electrolytes, capturing critical processes over extended time scales. Furthermore, we reveal the role of interface reaction kinetics in interface regulation through HAML simulations, combined with the similarity analysis method. It is demonstrated that element (Se, F, O) doping in the Li6PS5Cl system is an effective strategy for enhancing interface reaction kinetics, facilitating the formation of a more stable interface protective layer faster at room temperature. Moreover, moderate structural instability can positively contribute to interface stabilization. HAML offers a promising approach for addressing the challenge of designing stable interfaces while reducing computational costs. This work provides valuable insights for advancing the understanding and optimization of interface behaviors in Li metal batteries.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


