一种新的多系统ercc1 -肝肾综合征:来自临床队列、分子发病机制和管理指南的见解。
IF 4.6
2区 生物学
Q2 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
摘要
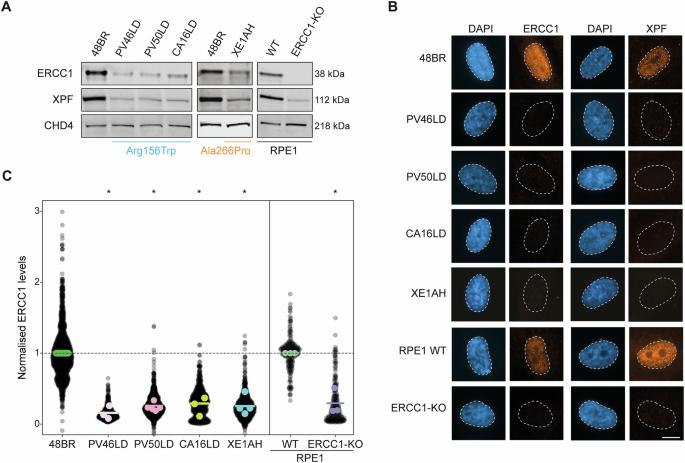
DNA修复障碍是一组以进行性多系统表型为特征的疾病。确定这些疾病的新临床表现对于优化患者护理至关重要。ERCC1-XPF是一种参与核苷酸切除修复(NER)和链间交联(ICL)修复的多功能内切酶。我们试图定义一种与双等位基因ERCC1变异和受损DNA修复相关的新型多系统表型。通过国际合作,我们从5个家族中鉴定出7名携带双等位基因ERCC1变异的个体,包括p.a g156trp和p.a ala266pro,他们表现出独特的临床表型。所有患者均表现为生长受限、光敏、肝肾功能障碍。值得注意的是,有三名儿童需要肝脏移植。四名儿童发生肝细胞癌,导致两人死亡,其中一人在接受阿霉素和顺铂治疗后死亡。老年人表现出其他特征,包括共济失调、基底细胞癌、胰腺功能不全、卵巢衰竭、甲状腺功能减退和限制性肺部疾病。使用患者来源的成纤维细胞进行的功能分析表明,ERCC1-XPF复合物具有显著的不稳定性,并且在NER和ICL修复中存在缺陷。然而,观察到残留的NER和ICL修复活性,表明错义变异体具有次型效应,它们要么存在于纯合子状态,要么存在于预测的功能缺失等位基因的反式状态。我们将ercc1 -肝肾综合征定义为一种严重的多系统DNA修复疾病,与高发病率和死亡率相关,包括儿童肝细胞癌的显著风险。我们建议管理指南强调癌症监测和化疗的谨慎,以尽量减少治疗相关的毒性。本文章由计算机程序翻译,如有差异,请以英文原文为准。
A new multisystem ERCC1-hepatorenal syndrome: insights from a clinical cohort, molecular pathogenesis, and management guidelines
DNA repair disorders are a group of conditions characterized by progressive, multisystem phenotypes. Defining new clinical presentations of these disorders is essential for optimizing patient care. ERCC1-XPF is a multifunctional endonuclease involved in nucleotide excision repair (NER) and interstrand crosslink (ICL) repair. We sought to define a novel multisystem phenotype associated with biallelic ERCC1 variants and impaired DNA repair. Through international collaboration, we identified seven individuals from five families carrying biallelic ERCC1 variants, including p.Arg156Trp and p.Ala266Pro, who exhibited a distinct clinical phenotype. All individuals presented with growth restriction, photosensitivity, and kidney and liver dysfunction. Notably, three children required liver transplants. Hepatocellular carcinoma developed in four children, resulting in two deaths, including one following treatment with doxorubicin and cisplatin. Older individuals exhibited additional features, including ataxia, basal cell carcinomas, pancreatic insufficiency, ovarian failure, hypothyroidism, and restrictive lung disease. Functional assays using patient-derived fibroblasts demonstrated significant destabilization of the ERCC1-XPF complex and defects in NER and ICL repair. However, residual NER and ICL repair activity was observed, suggesting a hypomorphic effect of the missense variants, which were present either in the homozygous state or in trans with a predicted loss-of-function allele. We define ERCC1-hepatorenal syndrome as a severe, multisystem DNA repair disorder associated with high morbidity and mortality, including a significant risk of pediatric hepatocellular carcinoma. We propose management guidelines emphasizing cancer surveillance and caution with chemotherapy to minimize treatment-related toxicity.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

European Journal of Human Genetics
生物-生化与分子生物学
CiteScore
9.90
自引率
5.80%
发文量
216
审稿时长
2 months
期刊介绍:
The European Journal of Human Genetics is the official journal of the European Society of Human Genetics, publishing high-quality, original research papers, short reports and reviews in the rapidly expanding field of human genetics and genomics. It covers molecular, clinical and cytogenetics, interfacing between advanced biomedical research and the clinician, and bridging the great diversity of facilities, resources and viewpoints in the genetics community.
Key areas include:
-Monogenic and multifactorial disorders
-Development and malformation
-Hereditary cancer
-Medical Genomics
-Gene mapping and functional studies
-Genotype-phenotype correlations
-Genetic variation and genome diversity
-Statistical and computational genetics
-Bioinformatics
-Advances in diagnostics
-Therapy and prevention
-Animal models
-Genetic services
-Community genetics
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


