可解释的机器学习框架:预测复合光催化剂对CO2转化的催化活性
IF 9.7
2区 材料科学
Q1 MATERIALS SCIENCE, MULTIDISCIPLINARY
引用次数: 0
摘要
CO2还原反应(CO2RR)的光催化活性受到中间体间吸附能的线性标度关系的限制。双原子光催化剂(DAPs)克服了这一限制,提高了催化效率。在这项工作中,我们开发了一个可解释的基于dft的机器学习(ML)框架,以预测和合理化299个基于过渡金属(TM)的DAPs锚定在化学计量当量石墨氮化碳(gC6N6)上的CO2RR活性。AdaBoost回归模型利用结构敏感(Magpie)和结构属性(Coulomb矩阵特征值)特征,准确预测潜在决定步长(PDS), R2 = 0.95, RMSE = 0.06 eV。特征选择发现固定的TM原子的平均电负性是最重要的描述符。ML模型显示ScTi/gC6N6和RhMo/gC6N6分别具有0.58 eV和0.73 eV的极限势,是很有前途的光催化剂。进一步的DFT计算证实了它们适合CO2RR的光电特性。这一策略能够实现准确和可解释的预测,潜在地加速了高效光催化材料的发现和设计。本文章由计算机程序翻译,如有差异,请以英文原文为准。

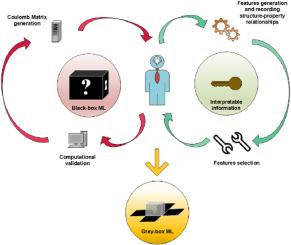
Interpretable machine learning framework: Predicting catalytic activities of complex photocatalysts towards CO2 conversion
The photocatalytic activity of CO2 conversion via reduction reaction (CO2RR) is limited by linear scaling relations of adsorption energies between intermediates. Dual-atom photocatalysts (DAPs) have been proposed to overcome this limitation and enhance catalytic efficiency. In this work, we develop an interpretable DFT-based machine learning (ML) framework to predict and rationalize the CO2RR activity of 299 transition metal (TM)-based DAPs anchored on stoichiometric equivalent graphitic carbon nitride (gC6N6). Leveraging structure-sensitive (Magpie) and structure-property (Coulomb Matrix Eigenvalues) features, the AdaBoost regressor model accurately predicts the potential determining step (PDS) with R2 = 0.95 and RMSE = 0.06 eV. Feature selection identified the mean electronegativity of the anchored TM atoms as the most important descriptor. The ML model highlights ScTi/gC6N6 and RhMo/gC6N6 as promising photocatalysts with limiting potentials of 0.58 eV and 0.73 eV, respectively. Further DFT calculations confirm their suitable optoelectronic properties for CO2RR. This strategy enables accurate and interpretable predictions, potentially accelerating the discovery and design of efficient photocatalytic materials.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Materials Today Physics
Materials Science-General Materials Science
CiteScore
14.00
自引率
7.80%
发文量
284
审稿时长
15 days
期刊介绍:
Materials Today Physics is a multi-disciplinary journal focused on the physics of materials, encompassing both the physical properties and materials synthesis. Operating at the interface of physics and materials science, this journal covers one of the largest and most dynamic fields within physical science. The forefront research in materials physics is driving advancements in new materials, uncovering new physics, and fostering novel applications at an unprecedented pace.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


