Zeng Liang, Kejiang Li, Jianliang Zhang, Alberto N. Conejo
{"title":"高温下非共沸物(Fe1-xO)中缺陷簇形成的启示:来自深度学习的精确力场","authors":"Zeng Liang, Kejiang Li, Jianliang Zhang, Alberto N. Conejo","doi":"10.1038/s41524-025-01527-3","DOIUrl":null,"url":null,"abstract":"<p>The limited understanding of the microstructure and dynamic evolution associated with the non-stoichiometric characteristics of wustite has constrained the comprehension of iron oxide properties, diffusion, and phase transformation behaviors. This study employs deep learning methods to train interatomic potential parameters for the Fe–O system, achieving precise atomic-scale simulations of the wustite phase structure and internal lattice defects. This approach addresses the shortcomings of large-scale molecular dynamics simulations in accurately describing the solid-phase structure of the Fe–O system. Utilizing these potential parameters, this research is the first to reveal the complex mechanisms underlying the non-stoichiometric nature of wustite (Fe<sub>1−<i>x</i></sub>O). The study found that cation vacancy defects in wustite tend to aggregate, forming stable cluster structures. It also elucidated the formation mechanisms of interstitial iron atoms and typical defect clusters in wustite, establishing the formation preference for Koch–Cohen defect clusters. These potential parameters and research methods can be further applied in future studies on iron oxide reduction, phase transformation mechanisms, and related material development, thereby advancing fundamental research in metallurgy and related industries.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"1 1","pages":""},"PeriodicalIF":9.4000,"publicationDate":"2025-02-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Insights into defect cluster formation in non-stoichiometric wustite (Fe1−xO) at elevated temperatures: accurate force field from deep learning\",\"authors\":\"Zeng Liang, Kejiang Li, Jianliang Zhang, Alberto N. Conejo\",\"doi\":\"10.1038/s41524-025-01527-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The limited understanding of the microstructure and dynamic evolution associated with the non-stoichiometric characteristics of wustite has constrained the comprehension of iron oxide properties, diffusion, and phase transformation behaviors. This study employs deep learning methods to train interatomic potential parameters for the Fe–O system, achieving precise atomic-scale simulations of the wustite phase structure and internal lattice defects. This approach addresses the shortcomings of large-scale molecular dynamics simulations in accurately describing the solid-phase structure of the Fe–O system. Utilizing these potential parameters, this research is the first to reveal the complex mechanisms underlying the non-stoichiometric nature of wustite (Fe<sub>1−<i>x</i></sub>O). The study found that cation vacancy defects in wustite tend to aggregate, forming stable cluster structures. It also elucidated the formation mechanisms of interstitial iron atoms and typical defect clusters in wustite, establishing the formation preference for Koch–Cohen defect clusters. These potential parameters and research methods can be further applied in future studies on iron oxide reduction, phase transformation mechanisms, and related material development, thereby advancing fundamental research in metallurgy and related industries.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"1 1\",\"pages\":\"\"},\"PeriodicalIF\":9.4000,\"publicationDate\":\"2025-02-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-025-01527-3\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-025-01527-3","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Insights into defect cluster formation in non-stoichiometric wustite (Fe1−xO) at elevated temperatures: accurate force field from deep learning
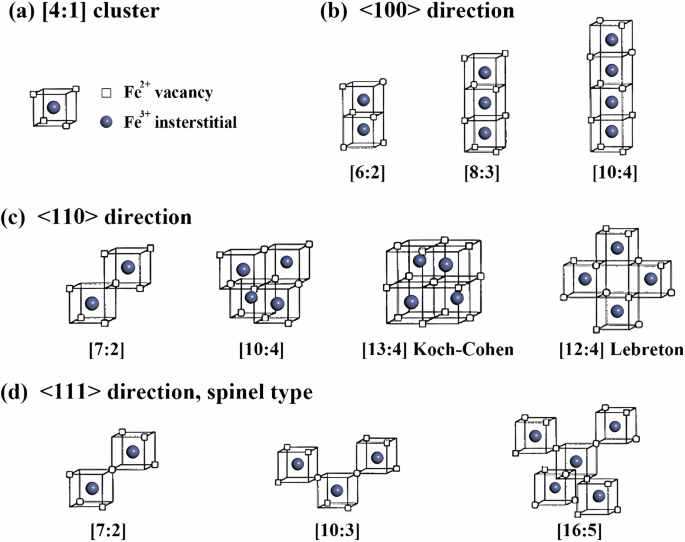
The limited understanding of the microstructure and dynamic evolution associated with the non-stoichiometric characteristics of wustite has constrained the comprehension of iron oxide properties, diffusion, and phase transformation behaviors. This study employs deep learning methods to train interatomic potential parameters for the Fe–O system, achieving precise atomic-scale simulations of the wustite phase structure and internal lattice defects. This approach addresses the shortcomings of large-scale molecular dynamics simulations in accurately describing the solid-phase structure of the Fe–O system. Utilizing these potential parameters, this research is the first to reveal the complex mechanisms underlying the non-stoichiometric nature of wustite (Fe1−xO). The study found that cation vacancy defects in wustite tend to aggregate, forming stable cluster structures. It also elucidated the formation mechanisms of interstitial iron atoms and typical defect clusters in wustite, establishing the formation preference for Koch–Cohen defect clusters. These potential parameters and research methods can be further applied in future studies on iron oxide reduction, phase transformation mechanisms, and related material development, thereby advancing fundamental research in metallurgy and related industries.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


