{"title":"通过基于集合的虚拟筛选方法、生物学评价和分子动力学模拟,鉴定靶向 PI3Kα 的新型抑制剂","authors":"Hui Zhang, Hua-Zhao Qi, Ya-Juan Li, Xiu-Yun Shi, Mei-Ling Hu, Xiang-Long Chen, Yuan Li","doi":"10.1007/s10822-024-00580-2","DOIUrl":null,"url":null,"abstract":"<div><p>PIK3CA gene encoding PI3K p110α is one of the most frequently mutated and overexpressed in majority of human cancers. Development of potent and selective novel inhibitors targeting PI3Kα was considered as the most promising approaches for cancer treatment. In this investigation, a virtual screening platform for PI3Kα inhibitors was established by employing machine learning methods, pharmacophore modeling, and molecular docking approaches. 28 potential PI3Kα inhibitors with different scaffolds were selected from the databases with 295,024 compounds. Among the 28 hits, hit15 exhibited the best inhibitory effect against PI3Kα with IC<sub>50</sub> value less than 1.0 µM. The molecular dynamics simulation indicated that hit15 could stably bind to the active site of PI3Kα, interact with some residues by hydrophobic, electrostatic and hydrogen bonding interactions, and finally induced PI3Kα active pocket substantial conformation changes. Stable H-bond interactions were formed between hit15 and residues of Lys776, Asp810 and Asp933. The binding free energy of PI3Kα-hit15 was − 65.3 kJ/mol. The free energy decomposition indicated that key residues of Asp805, Ile848 and Ile932 contributed stronger energies to the binding free energy. The above results indicated that hit15 with novel scaffold was a potent PI3Kα inhibitor and considered as a promising candidate for further drug development to treat various cancers with PI3Kα over activated.</p><h3>Graphical Abstract</h3>\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"38 1","pages":""},"PeriodicalIF":3.1000,"publicationDate":"2024-11-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Identification of novel inhibitors targeting PI3Kα via ensemble-based virtual screening method, biological evaluation and molecular dynamics simulation\",\"authors\":\"Hui Zhang, Hua-Zhao Qi, Ya-Juan Li, Xiu-Yun Shi, Mei-Ling Hu, Xiang-Long Chen, Yuan Li\",\"doi\":\"10.1007/s10822-024-00580-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>PIK3CA gene encoding PI3K p110α is one of the most frequently mutated and overexpressed in majority of human cancers. Development of potent and selective novel inhibitors targeting PI3Kα was considered as the most promising approaches for cancer treatment. In this investigation, a virtual screening platform for PI3Kα inhibitors was established by employing machine learning methods, pharmacophore modeling, and molecular docking approaches. 28 potential PI3Kα inhibitors with different scaffolds were selected from the databases with 295,024 compounds. Among the 28 hits, hit15 exhibited the best inhibitory effect against PI3Kα with IC<sub>50</sub> value less than 1.0 µM. The molecular dynamics simulation indicated that hit15 could stably bind to the active site of PI3Kα, interact with some residues by hydrophobic, electrostatic and hydrogen bonding interactions, and finally induced PI3Kα active pocket substantial conformation changes. Stable H-bond interactions were formed between hit15 and residues of Lys776, Asp810 and Asp933. The binding free energy of PI3Kα-hit15 was − 65.3 kJ/mol. The free energy decomposition indicated that key residues of Asp805, Ile848 and Ile932 contributed stronger energies to the binding free energy. The above results indicated that hit15 with novel scaffold was a potent PI3Kα inhibitor and considered as a promising candidate for further drug development to treat various cancers with PI3Kα over activated.</p><h3>Graphical Abstract</h3>\\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>\",\"PeriodicalId\":621,\"journal\":{\"name\":\"Journal of Computer-Aided Molecular Design\",\"volume\":\"38 1\",\"pages\":\"\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-11-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computer-Aided Molecular Design\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10822-024-00580-2\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-024-00580-2","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Identification of novel inhibitors targeting PI3Kα via ensemble-based virtual screening method, biological evaluation and molecular dynamics simulation
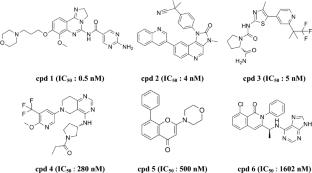
PIK3CA gene encoding PI3K p110α is one of the most frequently mutated and overexpressed in majority of human cancers. Development of potent and selective novel inhibitors targeting PI3Kα was considered as the most promising approaches for cancer treatment. In this investigation, a virtual screening platform for PI3Kα inhibitors was established by employing machine learning methods, pharmacophore modeling, and molecular docking approaches. 28 potential PI3Kα inhibitors with different scaffolds were selected from the databases with 295,024 compounds. Among the 28 hits, hit15 exhibited the best inhibitory effect against PI3Kα with IC50 value less than 1.0 µM. The molecular dynamics simulation indicated that hit15 could stably bind to the active site of PI3Kα, interact with some residues by hydrophobic, electrostatic and hydrogen bonding interactions, and finally induced PI3Kα active pocket substantial conformation changes. Stable H-bond interactions were formed between hit15 and residues of Lys776, Asp810 and Asp933. The binding free energy of PI3Kα-hit15 was − 65.3 kJ/mol. The free energy decomposition indicated that key residues of Asp805, Ile848 and Ile932 contributed stronger energies to the binding free energy. The above results indicated that hit15 with novel scaffold was a potent PI3Kα inhibitor and considered as a promising candidate for further drug development to treat various cancers with PI3Kα over activated.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


