Hoje Chun, Jaclyn R. Lunger, Jeung Ku Kang, Rafael Gómez-Bombarelli, Byungchan Han
{"title":"主动学习加速探索氧电催化多金属体系中的单原子局部环境","authors":"Hoje Chun, Jaclyn R. Lunger, Jeung Ku Kang, Rafael Gómez-Bombarelli, Byungchan Han","doi":"10.1038/s41524-024-01432-1","DOIUrl":null,"url":null,"abstract":"<p>Single-atom catalysts (SACs) with multiple active sites exhibit high activity for a wide range of sluggish reactions, but identifying optimal multimetallic SAC is challenging due to the vast design space. Here, we present a self-driving computational strategy that combines first-principles calculations and equivariant graph neural network (GNN) to explore over 30,000 binary metallic sites with varying combinations of 3<i>d</i> transition metals and different ligand environments for oxygen reduction and evolution reactions (ORR/OER). Active learning facilitates the investigation of the search space by balancing the exploration of unseen atomic structures with the exploitation of the active ones. The GNN learns the chemical environments to capture composition-structure-property relationships for ORR/OER activity and selectivity. The computational predictions of promising Co-Fe, Co-Co, and Co-Zn metal pairs are consistent with the state-of-the-art results of experimental measurements reported in the literature. This approach can be extended to a broader class of multi-element high entropic materials systems.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"8 1","pages":""},"PeriodicalIF":9.4000,"publicationDate":"2024-10-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Active learning accelerated exploration of single-atom local environments in multimetallic systems for oxygen electrocatalysis\",\"authors\":\"Hoje Chun, Jaclyn R. Lunger, Jeung Ku Kang, Rafael Gómez-Bombarelli, Byungchan Han\",\"doi\":\"10.1038/s41524-024-01432-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Single-atom catalysts (SACs) with multiple active sites exhibit high activity for a wide range of sluggish reactions, but identifying optimal multimetallic SAC is challenging due to the vast design space. Here, we present a self-driving computational strategy that combines first-principles calculations and equivariant graph neural network (GNN) to explore over 30,000 binary metallic sites with varying combinations of 3<i>d</i> transition metals and different ligand environments for oxygen reduction and evolution reactions (ORR/OER). Active learning facilitates the investigation of the search space by balancing the exploration of unseen atomic structures with the exploitation of the active ones. The GNN learns the chemical environments to capture composition-structure-property relationships for ORR/OER activity and selectivity. The computational predictions of promising Co-Fe, Co-Co, and Co-Zn metal pairs are consistent with the state-of-the-art results of experimental measurements reported in the literature. This approach can be extended to a broader class of multi-element high entropic materials systems.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"8 1\",\"pages\":\"\"},\"PeriodicalIF\":9.4000,\"publicationDate\":\"2024-10-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-024-01432-1\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01432-1","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Active learning accelerated exploration of single-atom local environments in multimetallic systems for oxygen electrocatalysis
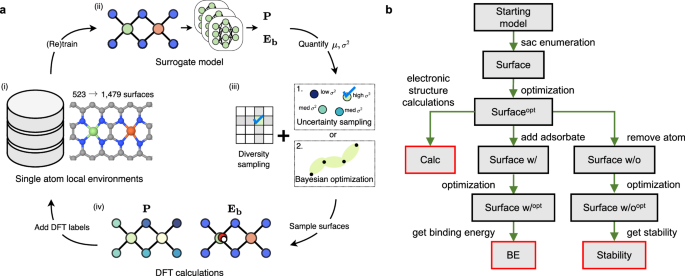
Single-atom catalysts (SACs) with multiple active sites exhibit high activity for a wide range of sluggish reactions, but identifying optimal multimetallic SAC is challenging due to the vast design space. Here, we present a self-driving computational strategy that combines first-principles calculations and equivariant graph neural network (GNN) to explore over 30,000 binary metallic sites with varying combinations of 3d transition metals and different ligand environments for oxygen reduction and evolution reactions (ORR/OER). Active learning facilitates the investigation of the search space by balancing the exploration of unseen atomic structures with the exploitation of the active ones. The GNN learns the chemical environments to capture composition-structure-property relationships for ORR/OER activity and selectivity. The computational predictions of promising Co-Fe, Co-Co, and Co-Zn metal pairs are consistent with the state-of-the-art results of experimental measurements reported in the literature. This approach can be extended to a broader class of multi-element high entropic materials systems.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


