{"title":"快速预测复杂有机分子的非谐波振动光谱","authors":"Mattia Miotto, Lorenzo Monacelli","doi":"10.1038/s41524-024-01400-9","DOIUrl":null,"url":null,"abstract":"<p>Interpreting Raman and IR vibrational spectra in complex organic molecules lacking symmetries poses a formidable challenge. In this study, we propose an innovative approach for simulating vibrational spectra and attributing observed peaks to molecular motions, even when highly anharmonic, without the need for computationally expensive ab initio calculations. Our approach stems from the time-dependent stochastic self-consistent harmonic approximation to capture quantum nuclear fluctuations in atom dynamics while describing interatomic interaction through state-of-the-art reactive machine-learning force fields. Finally, we employ an isotropic charge model and a bond capacitor model trained on ab initio data to predict the intensity of IR and Raman signals.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"62 1","pages":""},"PeriodicalIF":9.4000,"publicationDate":"2024-10-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Fast prediction of anharmonic vibrational spectra for complex organic molecules\",\"authors\":\"Mattia Miotto, Lorenzo Monacelli\",\"doi\":\"10.1038/s41524-024-01400-9\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Interpreting Raman and IR vibrational spectra in complex organic molecules lacking symmetries poses a formidable challenge. In this study, we propose an innovative approach for simulating vibrational spectra and attributing observed peaks to molecular motions, even when highly anharmonic, without the need for computationally expensive ab initio calculations. Our approach stems from the time-dependent stochastic self-consistent harmonic approximation to capture quantum nuclear fluctuations in atom dynamics while describing interatomic interaction through state-of-the-art reactive machine-learning force fields. Finally, we employ an isotropic charge model and a bond capacitor model trained on ab initio data to predict the intensity of IR and Raman signals.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"62 1\",\"pages\":\"\"},\"PeriodicalIF\":9.4000,\"publicationDate\":\"2024-10-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-024-01400-9\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01400-9","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
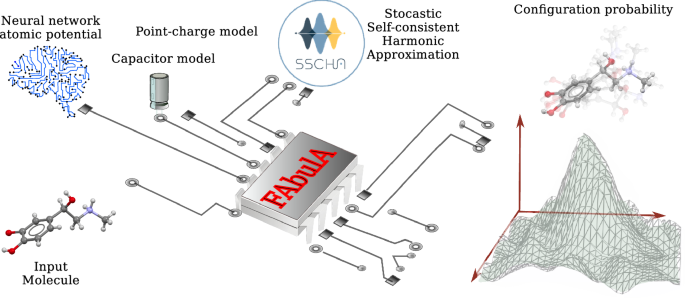
解读缺乏对称性的复杂有机分子的拉曼和红外振动光谱是一项艰巨的挑战。在本研究中,我们提出了一种创新方法,用于模拟振动光谱并将观测到的峰值归因于分子运动,即使是在高度非谐波的情况下,也无需进行计算成本高昂的 ab initio 计算。我们的方法源于随时间变化的随机自洽谐波近似,以捕捉原子动力学中的量子核波动,同时通过最先进的反应式机器学习力场来描述原子间的相互作用。最后,我们采用各向同性电荷模型和在 ab initio 数据基础上训练的键电容模型来预测红外和拉曼信号的强度。
Fast prediction of anharmonic vibrational spectra for complex organic molecules
Interpreting Raman and IR vibrational spectra in complex organic molecules lacking symmetries poses a formidable challenge. In this study, we propose an innovative approach for simulating vibrational spectra and attributing observed peaks to molecular motions, even when highly anharmonic, without the need for computationally expensive ab initio calculations. Our approach stems from the time-dependent stochastic self-consistent harmonic approximation to capture quantum nuclear fluctuations in atom dynamics while describing interatomic interaction through state-of-the-art reactive machine-learning force fields. Finally, we employ an isotropic charge model and a bond capacitor model trained on ab initio data to predict the intensity of IR and Raman signals.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


