{"title":"绘制富勒烯系统的结构-性能关系图:从 C20 到 C60 的计算研究","authors":"Bin Liu, Jirui Jin, Mingjie Liu","doi":"10.1038/s41524-024-01410-7","DOIUrl":null,"url":null,"abstract":"<p>Fullerenes, as characteristic carbon nanomaterials, offer significant potential for diverse applications due to their structural diversity and tunable properties. Numerous isomers can exist for a specific fullerene size, yet a comprehensive understanding of their fundamental properties remains elusive. In this study, we construct an up-to-date computational database for C<sub>20</sub>–C<sub>60</sub> fullerenes, consisting of 5770 structures, and calculate 12 fundamental properties using DFT, including stability (binding energy), electronic properties (HOMO-LUMO gap), and solubility (partition coefficient logP). Our findings reveal that the HOMO-LUMO gap weakly correlates with both binding energy and logP, indicating that electronic properties can be tailored for specific uses without affecting stability or solubility. In addition, we introduce a set of topological features and geometric measures to investigate structure-property relationships. We apply atom, bond, and hexagon features to effectively predict the stability of C<sub>20</sub>–C<sub>60</sub> fullerenes, surpassing the conventional qualitative isolated pentagon rule, and demonstrating their robust transferability to larger-size fullerenes beyond C<sub>60</sub>. Our work offers guidance for optimizing fullerenes as electron acceptors in organic solar cells and lays a foundational understanding of their functionalization and applications in energy conversion and nanomaterial sciences.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"38 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2024-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Mapping structure-property relationships in fullerene systems: a computational study from C20 to C60\",\"authors\":\"Bin Liu, Jirui Jin, Mingjie Liu\",\"doi\":\"10.1038/s41524-024-01410-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Fullerenes, as characteristic carbon nanomaterials, offer significant potential for diverse applications due to their structural diversity and tunable properties. Numerous isomers can exist for a specific fullerene size, yet a comprehensive understanding of their fundamental properties remains elusive. In this study, we construct an up-to-date computational database for C<sub>20</sub>–C<sub>60</sub> fullerenes, consisting of 5770 structures, and calculate 12 fundamental properties using DFT, including stability (binding energy), electronic properties (HOMO-LUMO gap), and solubility (partition coefficient logP). Our findings reveal that the HOMO-LUMO gap weakly correlates with both binding energy and logP, indicating that electronic properties can be tailored for specific uses without affecting stability or solubility. In addition, we introduce a set of topological features and geometric measures to investigate structure-property relationships. We apply atom, bond, and hexagon features to effectively predict the stability of C<sub>20</sub>–C<sub>60</sub> fullerenes, surpassing the conventional qualitative isolated pentagon rule, and demonstrating their robust transferability to larger-size fullerenes beyond C<sub>60</sub>. Our work offers guidance for optimizing fullerenes as electron acceptors in organic solar cells and lays a foundational understanding of their functionalization and applications in energy conversion and nanomaterial sciences.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"38 1\",\"pages\":\"\"},\"PeriodicalIF\":11.9000,\"publicationDate\":\"2024-09-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-024-01410-7\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01410-7","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Mapping structure-property relationships in fullerene systems: a computational study from C20 to C60
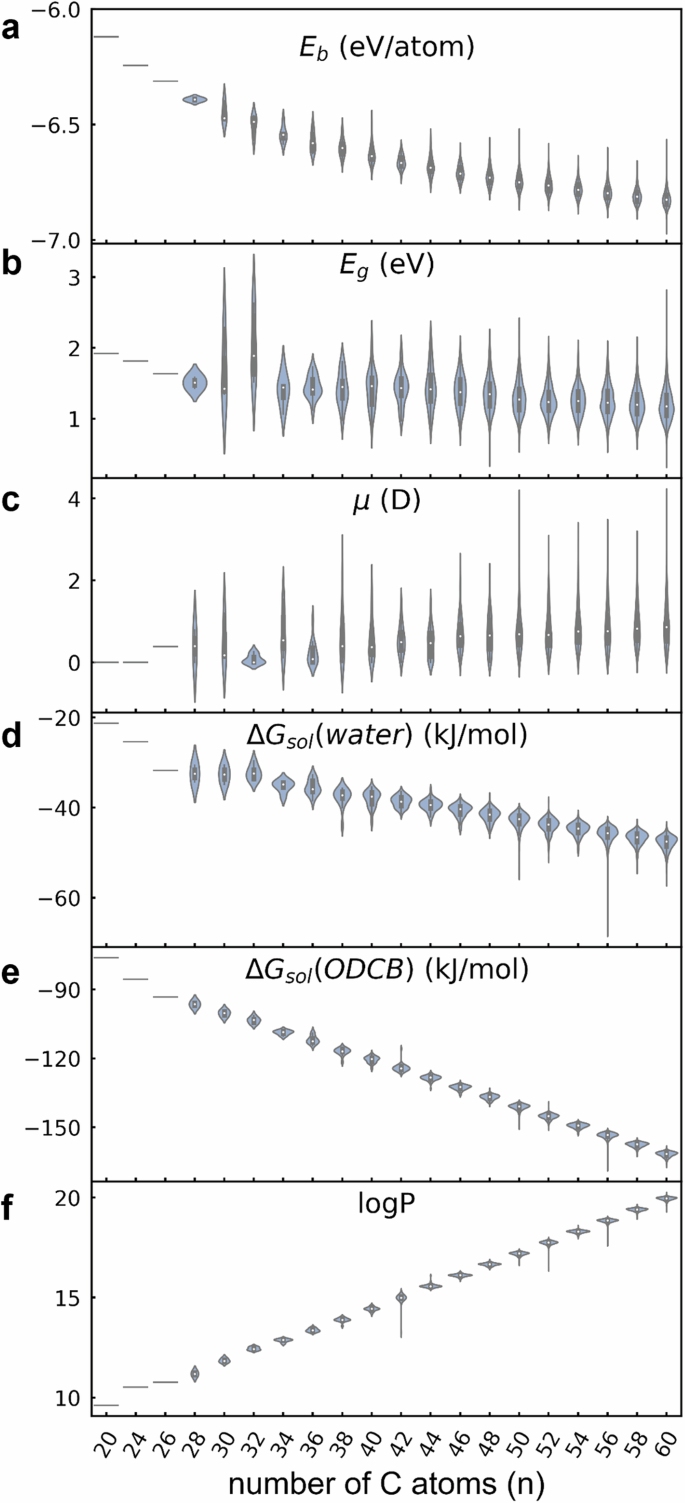
Fullerenes, as characteristic carbon nanomaterials, offer significant potential for diverse applications due to their structural diversity and tunable properties. Numerous isomers can exist for a specific fullerene size, yet a comprehensive understanding of their fundamental properties remains elusive. In this study, we construct an up-to-date computational database for C20–C60 fullerenes, consisting of 5770 structures, and calculate 12 fundamental properties using DFT, including stability (binding energy), electronic properties (HOMO-LUMO gap), and solubility (partition coefficient logP). Our findings reveal that the HOMO-LUMO gap weakly correlates with both binding energy and logP, indicating that electronic properties can be tailored for specific uses without affecting stability or solubility. In addition, we introduce a set of topological features and geometric measures to investigate structure-property relationships. We apply atom, bond, and hexagon features to effectively predict the stability of C20–C60 fullerenes, surpassing the conventional qualitative isolated pentagon rule, and demonstrating their robust transferability to larger-size fullerenes beyond C60. Our work offers guidance for optimizing fullerenes as electron acceptors in organic solar cells and lays a foundational understanding of their functionalization and applications in energy conversion and nanomaterial sciences.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


