{"title":"用 E(3)等变图神经网络表征短程秩序的化学特征","authors":"Killian Sheriff, Yifan Cao, Rodrigo Freitas","doi":"10.1038/s41524-024-01393-5","DOIUrl":null,"url":null,"abstract":"<p>Crystalline materials have atomic-scale fluctuations in their chemical composition that modulate various mesoscale properties. Establishing chemistry–microstructure relationships in such materials requires proper characterization of these chemical fluctuations. Yet, current characterization approaches (e.g., Warren–Cowley parameters) make only partial use of the complete chemical and structural information contained in local chemical motifs. Here we introduce a framework based on E(3)-equivariant graph neural networks that is capable of completely identifying chemical motifs in arbitrary crystalline structures with any number of chemical elements. This approach naturally leads to a proper information-theoretic measure for quantifying chemical short-range order (SRO) in chemically complex materials and a reduced representation of the chemical motif space. Our framework enables the correlation of any per-atom property with their corresponding local chemical motif, thereby enabling the exploration of structure–property relationships in chemically complex materials. Using the MoTaNbTi high-entropy alloy as a test system, we demonstrate the versatility of this approach by evaluating the lattice strain associated with each chemical motif, and computing the temperature dependence of chemical-fluctuations length scale.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"10 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2024-09-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Chemical-motif characterization of short-range order with E(3)-equivariant graph neural networks\",\"authors\":\"Killian Sheriff, Yifan Cao, Rodrigo Freitas\",\"doi\":\"10.1038/s41524-024-01393-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Crystalline materials have atomic-scale fluctuations in their chemical composition that modulate various mesoscale properties. Establishing chemistry–microstructure relationships in such materials requires proper characterization of these chemical fluctuations. Yet, current characterization approaches (e.g., Warren–Cowley parameters) make only partial use of the complete chemical and structural information contained in local chemical motifs. Here we introduce a framework based on E(3)-equivariant graph neural networks that is capable of completely identifying chemical motifs in arbitrary crystalline structures with any number of chemical elements. This approach naturally leads to a proper information-theoretic measure for quantifying chemical short-range order (SRO) in chemically complex materials and a reduced representation of the chemical motif space. Our framework enables the correlation of any per-atom property with their corresponding local chemical motif, thereby enabling the exploration of structure–property relationships in chemically complex materials. Using the MoTaNbTi high-entropy alloy as a test system, we demonstrate the versatility of this approach by evaluating the lattice strain associated with each chemical motif, and computing the temperature dependence of chemical-fluctuations length scale.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"10 1\",\"pages\":\"\"},\"PeriodicalIF\":11.9000,\"publicationDate\":\"2024-09-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-024-01393-5\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01393-5","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Chemical-motif characterization of short-range order with E(3)-equivariant graph neural networks
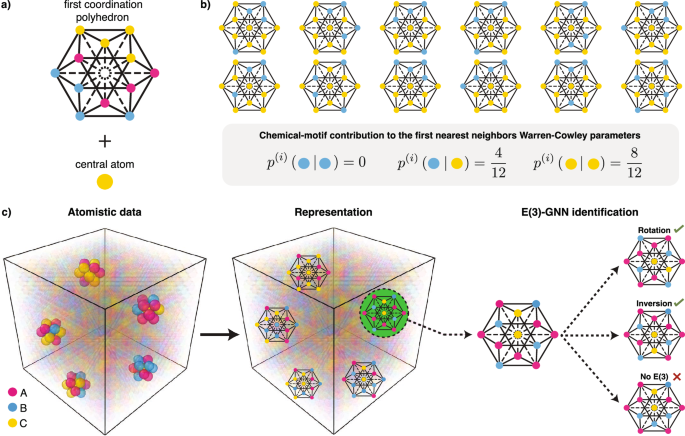
Crystalline materials have atomic-scale fluctuations in their chemical composition that modulate various mesoscale properties. Establishing chemistry–microstructure relationships in such materials requires proper characterization of these chemical fluctuations. Yet, current characterization approaches (e.g., Warren–Cowley parameters) make only partial use of the complete chemical and structural information contained in local chemical motifs. Here we introduce a framework based on E(3)-equivariant graph neural networks that is capable of completely identifying chemical motifs in arbitrary crystalline structures with any number of chemical elements. This approach naturally leads to a proper information-theoretic measure for quantifying chemical short-range order (SRO) in chemically complex materials and a reduced representation of the chemical motif space. Our framework enables the correlation of any per-atom property with their corresponding local chemical motif, thereby enabling the exploration of structure–property relationships in chemically complex materials. Using the MoTaNbTi high-entropy alloy as a test system, we demonstrate the versatility of this approach by evaluating the lattice strain associated with each chemical motif, and computing the temperature dependence of chemical-fluctuations length scale.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:



