Brian H. Lee, James P. Larentzos, John K. Brennan, Alejandro Strachan
{"title":"分子晶体 RDX 的图神经网络粗粒力场","authors":"Brian H. Lee, James P. Larentzos, John K. Brennan, Alejandro Strachan","doi":"10.1038/s41524-024-01407-2","DOIUrl":null,"url":null,"abstract":"<p>Condense phase molecular systems organize in wide range of distinct molecular configurations, including amorphous melt and glass as well as crystals often exhibiting polymorphism, that originate from their intricate intra- and intermolecular forces. While accurate coarse-grain (CG) models for these materials are critical to understand phenomena beyond the reach of all-atom simulations, current models cannot capture the diversity of molecular structures. We introduce a generally applicable approach to develop CG force fields for molecular crystals combining graph neural networks (GNN) and data from an all-atom simulations and apply it to the high-energy density material RDX. We address the challenge of expanding the training data with relevant configurations via an iterative procedure that performs CG molecular dynamics of processes of interest and reconstructs the atomistic configurations using a pre-trained neural network decoder. The multi-site CG model uses a GNN architecture constructed to satisfy translational invariance and rotational covariance for forces. The resulting model captures both crystalline and amorphous states for a wide range of temperatures and densities.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"46 1","pages":""},"PeriodicalIF":9.4000,"publicationDate":"2024-09-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Graph neural network coarse-grain force field for the molecular crystal RDX\",\"authors\":\"Brian H. Lee, James P. Larentzos, John K. Brennan, Alejandro Strachan\",\"doi\":\"10.1038/s41524-024-01407-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Condense phase molecular systems organize in wide range of distinct molecular configurations, including amorphous melt and glass as well as crystals often exhibiting polymorphism, that originate from their intricate intra- and intermolecular forces. While accurate coarse-grain (CG) models for these materials are critical to understand phenomena beyond the reach of all-atom simulations, current models cannot capture the diversity of molecular structures. We introduce a generally applicable approach to develop CG force fields for molecular crystals combining graph neural networks (GNN) and data from an all-atom simulations and apply it to the high-energy density material RDX. We address the challenge of expanding the training data with relevant configurations via an iterative procedure that performs CG molecular dynamics of processes of interest and reconstructs the atomistic configurations using a pre-trained neural network decoder. The multi-site CG model uses a GNN architecture constructed to satisfy translational invariance and rotational covariance for forces. The resulting model captures both crystalline and amorphous states for a wide range of temperatures and densities.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"46 1\",\"pages\":\"\"},\"PeriodicalIF\":9.4000,\"publicationDate\":\"2024-09-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-024-01407-2\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01407-2","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Graph neural network coarse-grain force field for the molecular crystal RDX
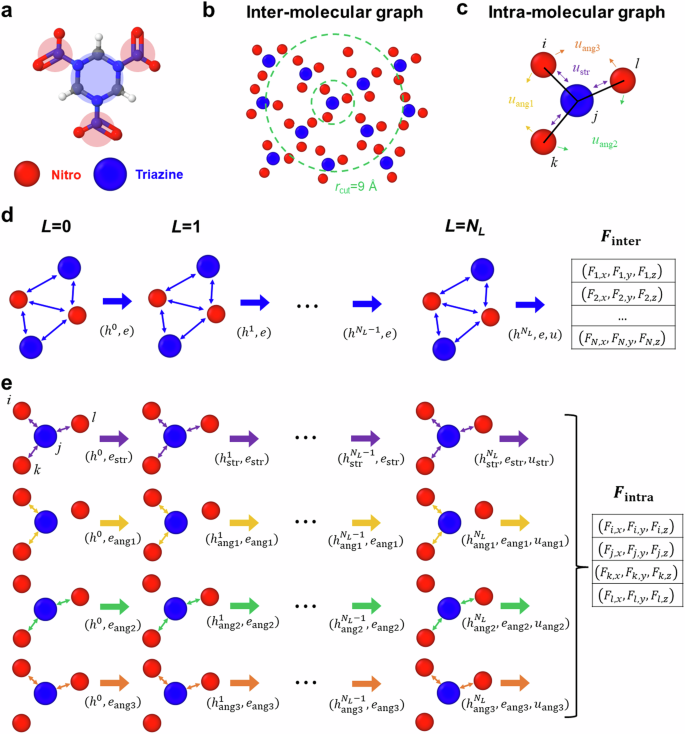
Condense phase molecular systems organize in wide range of distinct molecular configurations, including amorphous melt and glass as well as crystals often exhibiting polymorphism, that originate from their intricate intra- and intermolecular forces. While accurate coarse-grain (CG) models for these materials are critical to understand phenomena beyond the reach of all-atom simulations, current models cannot capture the diversity of molecular structures. We introduce a generally applicable approach to develop CG force fields for molecular crystals combining graph neural networks (GNN) and data from an all-atom simulations and apply it to the high-energy density material RDX. We address the challenge of expanding the training data with relevant configurations via an iterative procedure that performs CG molecular dynamics of processes of interest and reconstructs the atomistic configurations using a pre-trained neural network decoder. The multi-site CG model uses a GNN architecture constructed to satisfy translational invariance and rotational covariance for forces. The resulting model captures both crystalline and amorphous states for a wide range of temperatures and densities.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


