{"title":"通过不确定性量化实现迁移学习,预测数百万原子系统的电子结构","authors":"Shashank Pathrudkar, Ponkrshnan Thiagarajan, Shivang Agarwal, Amartya S. Banerjee, Susanta Ghosh","doi":"10.1038/s41524-024-01305-7","DOIUrl":null,"url":null,"abstract":"<p>The ground state electron density — obtainable using Kohn-Sham Density Functional Theory (KS-DFT) simulations — contains a wealth of material information, making its prediction via machine learning (ML) models attractive. However, the computational expense of KS-DFT scales cubically with system size which tends to stymie training data generation, making it difficult to develop quantifiably accurate ML models that are applicable across many scales and system configurations. Here, we address this fundamental challenge by employing transfer learning to leverage the multi-scale nature of the training data, while comprehensively sampling system configurations using thermalization. Our ML models are less reliant on heuristics, and being based on Bayesian neural networks, enable uncertainty quantification. We show that our models incur significantly lower data generation costs while allowing confident — and when verifiable, accurate — predictions for a wide variety of bulk systems well beyond training, including systems with defects, different alloy compositions, and at multi-million-atom scales. Moreover, such predictions can be carried out using only modest computational resources.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"74 1","pages":""},"PeriodicalIF":9.4000,"publicationDate":"2024-08-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Electronic structure prediction of multi-million atom systems through uncertainty quantification enabled transfer learning\",\"authors\":\"Shashank Pathrudkar, Ponkrshnan Thiagarajan, Shivang Agarwal, Amartya S. Banerjee, Susanta Ghosh\",\"doi\":\"10.1038/s41524-024-01305-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The ground state electron density — obtainable using Kohn-Sham Density Functional Theory (KS-DFT) simulations — contains a wealth of material information, making its prediction via machine learning (ML) models attractive. However, the computational expense of KS-DFT scales cubically with system size which tends to stymie training data generation, making it difficult to develop quantifiably accurate ML models that are applicable across many scales and system configurations. Here, we address this fundamental challenge by employing transfer learning to leverage the multi-scale nature of the training data, while comprehensively sampling system configurations using thermalization. Our ML models are less reliant on heuristics, and being based on Bayesian neural networks, enable uncertainty quantification. We show that our models incur significantly lower data generation costs while allowing confident — and when verifiable, accurate — predictions for a wide variety of bulk systems well beyond training, including systems with defects, different alloy compositions, and at multi-million-atom scales. Moreover, such predictions can be carried out using only modest computational resources.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"74 1\",\"pages\":\"\"},\"PeriodicalIF\":9.4000,\"publicationDate\":\"2024-08-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-024-01305-7\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01305-7","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
基态电子密度--可通过 Kohn-Sham 密度功能理论(KS-DFT)模拟获得--包含丰富的材料信息,因此通过机器学习(ML)模型对其进行预测极具吸引力。然而,KS-DFT 的计算费用与系统规模成三次方关系,往往会阻碍训练数据的生成,因此很难开发出适用于多种规模和系统配置的可量化的精确 ML 模型。在这里,我们采用迁移学习来利用训练数据的多尺度性质,同时利用热化对系统配置进行全面采样,从而解决了这一根本性挑战。我们的 ML 模型较少依赖启发式方法,并且基于贝叶斯神经网络,能够量化不确定性。我们的研究表明,我们的模型大大降低了数据生成成本,同时还能对各种散装系统(包括有缺陷的系统、不同的合金成分和数百万原子尺度的系统)进行有把握的预测,而且在可验证的情况下,预测结果也非常准确。此外,此类预测只需少量计算资源即可完成。
Electronic structure prediction of multi-million atom systems through uncertainty quantification enabled transfer learning
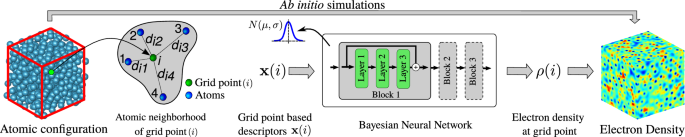
The ground state electron density — obtainable using Kohn-Sham Density Functional Theory (KS-DFT) simulations — contains a wealth of material information, making its prediction via machine learning (ML) models attractive. However, the computational expense of KS-DFT scales cubically with system size which tends to stymie training data generation, making it difficult to develop quantifiably accurate ML models that are applicable across many scales and system configurations. Here, we address this fundamental challenge by employing transfer learning to leverage the multi-scale nature of the training data, while comprehensively sampling system configurations using thermalization. Our ML models are less reliant on heuristics, and being based on Bayesian neural networks, enable uncertainty quantification. We show that our models incur significantly lower data generation costs while allowing confident — and when verifiable, accurate — predictions for a wide variety of bulk systems well beyond training, including systems with defects, different alloy compositions, and at multi-million-atom scales. Moreover, such predictions can be carried out using only modest computational resources.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


