评估 AlphaMissense 识别影响常见疾病易感性变异的能力。
IF 4.6
2区 生物学
Q2 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
摘要
罕见变异关联研究分析中的一个重要问题是,能否根据非同义变异影响蛋白质功能的重要性对其进行注释。为了解决这个问题,AlphaMissense 最近发布了,根据导致严重疾病的变异和功能测试的基准,它被证明具有良好的性能。在这里,我们评估了 AlphaMissense 在 18 个基因中的表现,这些基因以前曾被证明与罕见编码变异和高脂血症、高血压或 2 型糖尿病有关。我们比较了 AlphaMissense 和其他 43 种注释方法之间以符号对数值(SLP)表示的支持关联的证据强度。结果表明,在非同义变异对关联证据的贡献程度方面,不同基因之间存在明显差异,不同非同义变异注释方法的性能之间也存在明显差异。虽然 AlphaMissense 在各基因中平均产生了最高的 SLP,但它只对 4 个基因产生了最大的 SLP。对于某些基因,其他方法产生的 SLP 要高得多,还有一些基因,AlphaMissense 没有产生关联的证据,而另一种方法却有很好的表现。不同基因之间存在明显的不一致性,这意味着很难确定分析序列数据的最佳方法。不同的方法对不同的基因都有很好的效果,这一事实表明,如果希望使用序列数据进行个体风险预测,就应该使用特定基因的注释方法。本文章由计算机程序翻译,如有差异,请以英文原文为准。
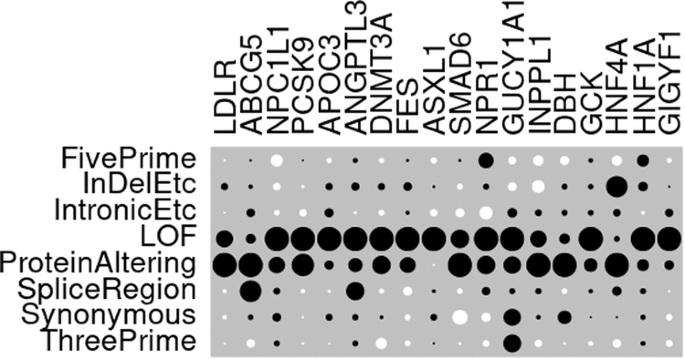
Assessment of ability of AlphaMissense to identify variants affecting susceptibility to common disease
An important issue in the analysis of rare variant association studies is the ability to annotate nonsynonymous variants in terms of their likely importance as affecting protein function. To address this, AlphaMissense was recently released and was shown to have good performance using benchmarks based on variants causing severe disease and on functional assays. Here, we assess the performance of AlphaMissense across 18 genes which had previously demonstrated association between rare coding variants and hyperlipidaemia, hypertension or type 2 diabetes. The strength of evidence in favour of association, expressed as the signed log p value (SLP), was compared between AlphaMissense and 43 other annotation methods. The results demonstrated marked variability between genes regarding the extent to which nonsynonymous variants contributed to evidence for association and also between the performance of different methods of annotating the nonsynonymous variants. Although AlphaMissense produced the highest SLP on average across genes, it produced the maximum SLP for only 4 genes. For some genes, other methods produced a considerably higher SLP and there were examples of genes where AlphaMissense produced no evidence for association while another method performed well. The marked inconsistency across genes means that it is difficult to decide on an optimal method of analysis of sequence data. The fact that different methods perform well for different genes suggests that if one wished to use sequence data for individual risk prediction then gene-specific annotation methods should be used.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

European Journal of Human Genetics
生物-生化与分子生物学
CiteScore
9.90
自引率
5.80%
发文量
216
审稿时长
2 months
期刊介绍:
The European Journal of Human Genetics is the official journal of the European Society of Human Genetics, publishing high-quality, original research papers, short reports and reviews in the rapidly expanding field of human genetics and genomics. It covers molecular, clinical and cytogenetics, interfacing between advanced biomedical research and the clinician, and bridging the great diversity of facilities, resources and viewpoints in the genetics community.
Key areas include:
-Monogenic and multifactorial disorders
-Development and malformation
-Hereditary cancer
-Medical Genomics
-Gene mapping and functional studies
-Genotype-phenotype correlations
-Genetic variation and genome diversity
-Statistical and computational genetics
-Bioinformatics
-Advances in diagnostics
-Therapy and prevention
-Animal models
-Genetic services
-Community genetics
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


