Wen-Chieh Huang, Chia-Hung Hsu, Titus V. Albu, Chia-Ning Yang
{"title":"两种与疾病相关的 ADAR1 突变体的结构影响:分子动力学研究。","authors":"Wen-Chieh Huang, Chia-Hung Hsu, Titus V. Albu, Chia-Ning Yang","doi":"10.1007/s10822-024-00565-1","DOIUrl":null,"url":null,"abstract":"<div><p>Adenosine deaminases acting on RNA (ADARs) are pivotal RNA-editing enzymes responsible for converting adenosine to inosine within double-stranded RNA (dsRNA). Dysregulation of ADAR1 editing activity, often arising from genetic mutations, has been linked to elevated interferon levels and the onset of autoinflammatory diseases. However, understanding the molecular underpinnings of this dysregulation is impeded by the lack of an experimentally determined structure for the ADAR1 deaminase domain. In this computational study, we utilized homology modeling and the AlphaFold2 to construct structural models of the ADAR1 deaminase domain in wild-type and two pathogenic variants, R892H and Y1112F, to decipher the structural impact on the reduced deaminase activity. Our findings illuminate the critical role of structural complementarity between the ADAR1 deaminase domain and dsRNA in enzyme-substrate recognition. That is, the relative position of E1008 and K1120 must be maintained so that they can insert into the minor and major grooves of the substrate dsRNA, respectively, facilitating the flipping-out of adenosine to be accommodated within a cavity surrounding E912. Both amino acid replacements studied, R892H at the orthosteric site and Y1112F at the allosteric site, alter K1120 position and ultimately hinder substrate RNA binding.</p></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"38 1","pages":""},"PeriodicalIF":3.0000,"publicationDate":"2024-07-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Structural impacts of two disease-linked ADAR1 mutants: a molecular dynamics study\",\"authors\":\"Wen-Chieh Huang, Chia-Hung Hsu, Titus V. Albu, Chia-Ning Yang\",\"doi\":\"10.1007/s10822-024-00565-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Adenosine deaminases acting on RNA (ADARs) are pivotal RNA-editing enzymes responsible for converting adenosine to inosine within double-stranded RNA (dsRNA). Dysregulation of ADAR1 editing activity, often arising from genetic mutations, has been linked to elevated interferon levels and the onset of autoinflammatory diseases. However, understanding the molecular underpinnings of this dysregulation is impeded by the lack of an experimentally determined structure for the ADAR1 deaminase domain. In this computational study, we utilized homology modeling and the AlphaFold2 to construct structural models of the ADAR1 deaminase domain in wild-type and two pathogenic variants, R892H and Y1112F, to decipher the structural impact on the reduced deaminase activity. Our findings illuminate the critical role of structural complementarity between the ADAR1 deaminase domain and dsRNA in enzyme-substrate recognition. That is, the relative position of E1008 and K1120 must be maintained so that they can insert into the minor and major grooves of the substrate dsRNA, respectively, facilitating the flipping-out of adenosine to be accommodated within a cavity surrounding E912. Both amino acid replacements studied, R892H at the orthosteric site and Y1112F at the allosteric site, alter K1120 position and ultimately hinder substrate RNA binding.</p></div>\",\"PeriodicalId\":621,\"journal\":{\"name\":\"Journal of Computer-Aided Molecular Design\",\"volume\":\"38 1\",\"pages\":\"\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-07-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computer-Aided Molecular Design\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10822-024-00565-1\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-024-00565-1","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
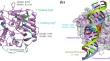
Structural impacts of two disease-linked ADAR1 mutants: a molecular dynamics study
Adenosine deaminases acting on RNA (ADARs) are pivotal RNA-editing enzymes responsible for converting adenosine to inosine within double-stranded RNA (dsRNA). Dysregulation of ADAR1 editing activity, often arising from genetic mutations, has been linked to elevated interferon levels and the onset of autoinflammatory diseases. However, understanding the molecular underpinnings of this dysregulation is impeded by the lack of an experimentally determined structure for the ADAR1 deaminase domain. In this computational study, we utilized homology modeling and the AlphaFold2 to construct structural models of the ADAR1 deaminase domain in wild-type and two pathogenic variants, R892H and Y1112F, to decipher the structural impact on the reduced deaminase activity. Our findings illuminate the critical role of structural complementarity between the ADAR1 deaminase domain and dsRNA in enzyme-substrate recognition. That is, the relative position of E1008 and K1120 must be maintained so that they can insert into the minor and major grooves of the substrate dsRNA, respectively, facilitating the flipping-out of adenosine to be accommodated within a cavity surrounding E912. Both amino acid replacements studied, R892H at the orthosteric site and Y1112F at the allosteric site, alter K1120 position and ultimately hinder substrate RNA binding.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


