{"title":"艾卡迪-古蒂耶尔综合征 1 型:一种新的错义变异和突变谱回顾。","authors":"Behnoosh Tasharrofi, Parvaneh Karimzadeh, Mostafa Asadollahi, Sepideh Hasani, Morteza Heidari, Mohammad Keramatipour","doi":"10.22037/ijcn.v18i3.43274","DOIUrl":null,"url":null,"abstract":"<p><strong>Objectives: </strong>Mutations in the TREX1 gene cause Aicardi-Goutières syndrome (AGS) 1, associated with a spectrum of autoimmune and neurodegenerative manifestations. AGS 1, the most severe neonatal type of AGS, is characterized by abnormal neurologic findings, visual inattention, hepatosplenomegaly, thrombocytopenia, skin rash, restlessness, and fever.</p><p><strong>Materials & methods: </strong>The present study described two affected siblings from an Iranian family whose phenotypes overlap with intrauterine infections. They had almost similar presentations, including developmental delay, microcephaly, no fix and follow epileptic seizures and the same pattern of brain CT scan involvements. Following clinical and paraclinical assessments, whole-exome sequencing was employed to determine the disease-causing variant, and subsequently, PCR-Sanger sequencing was performed to indicate the segregation pattern of the candidate variant in family members.</p><p><strong>Results: </strong>Genetic analysis revealed a novel homozygous missense variant (c.461A>C; p.D154A) in the TREX1 gene in affected family members. Sanger sequencing of other family members showed the expected zygosities.</p><p><strong>Conclusion: </strong>This study identifies a novel mutation in the TREX1 gene in this family and highlights the efficiency of next-generation sequencing-based techniques for obtaining a definite diagnosis in patients with early-onset encephalopathy.</p>","PeriodicalId":14537,"journal":{"name":"Iranian Journal of Child Neurology","volume":"18 3","pages":"117-129"},"PeriodicalIF":0.9000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11231675/pdf/","citationCount":"0","resultStr":"{\"title\":\"Aicardi-Goutières Syndrome Type 1: A Novel Missense Variant and Review of the Mutational Spectrum.\",\"authors\":\"Behnoosh Tasharrofi, Parvaneh Karimzadeh, Mostafa Asadollahi, Sepideh Hasani, Morteza Heidari, Mohammad Keramatipour\",\"doi\":\"10.22037/ijcn.v18i3.43274\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objectives: </strong>Mutations in the TREX1 gene cause Aicardi-Goutières syndrome (AGS) 1, associated with a spectrum of autoimmune and neurodegenerative manifestations. AGS 1, the most severe neonatal type of AGS, is characterized by abnormal neurologic findings, visual inattention, hepatosplenomegaly, thrombocytopenia, skin rash, restlessness, and fever.</p><p><strong>Materials & methods: </strong>The present study described two affected siblings from an Iranian family whose phenotypes overlap with intrauterine infections. They had almost similar presentations, including developmental delay, microcephaly, no fix and follow epileptic seizures and the same pattern of brain CT scan involvements. Following clinical and paraclinical assessments, whole-exome sequencing was employed to determine the disease-causing variant, and subsequently, PCR-Sanger sequencing was performed to indicate the segregation pattern of the candidate variant in family members.</p><p><strong>Results: </strong>Genetic analysis revealed a novel homozygous missense variant (c.461A>C; p.D154A) in the TREX1 gene in affected family members. Sanger sequencing of other family members showed the expected zygosities.</p><p><strong>Conclusion: </strong>This study identifies a novel mutation in the TREX1 gene in this family and highlights the efficiency of next-generation sequencing-based techniques for obtaining a definite diagnosis in patients with early-onset encephalopathy.</p>\",\"PeriodicalId\":14537,\"journal\":{\"name\":\"Iranian Journal of Child Neurology\",\"volume\":\"18 3\",\"pages\":\"117-129\"},\"PeriodicalIF\":0.9000,\"publicationDate\":\"2024-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11231675/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Iranian Journal of Child Neurology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.22037/ijcn.v18i3.43274\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/6/22 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Iranian Journal of Child Neurology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.22037/ijcn.v18i3.43274","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/6/22 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Aicardi-Goutières Syndrome Type 1: A Novel Missense Variant and Review of the Mutational Spectrum.
Objectives: Mutations in the TREX1 gene cause Aicardi-Goutières syndrome (AGS) 1, associated with a spectrum of autoimmune and neurodegenerative manifestations. AGS 1, the most severe neonatal type of AGS, is characterized by abnormal neurologic findings, visual inattention, hepatosplenomegaly, thrombocytopenia, skin rash, restlessness, and fever.
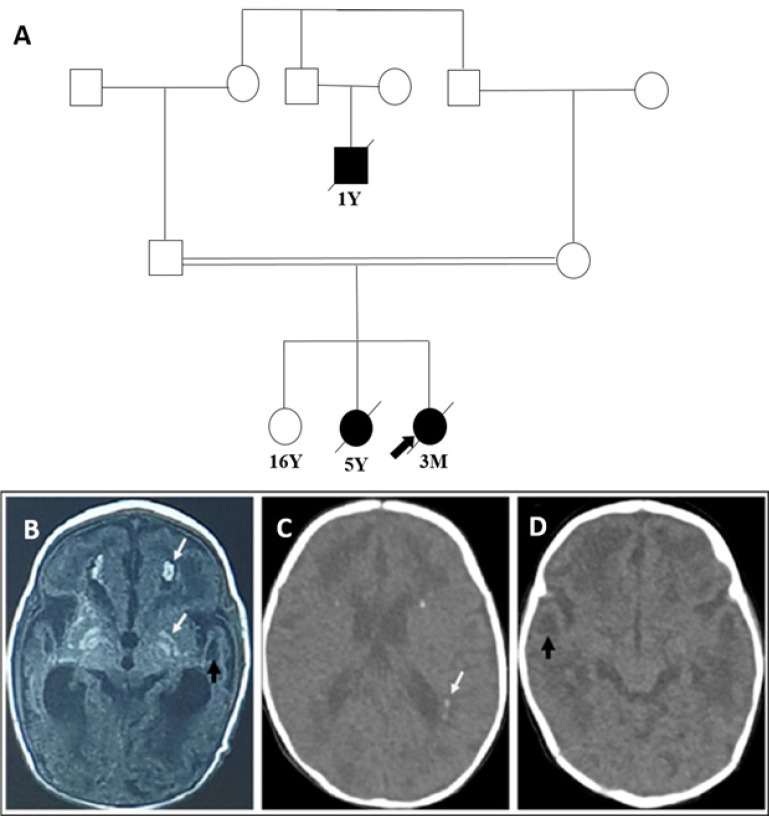
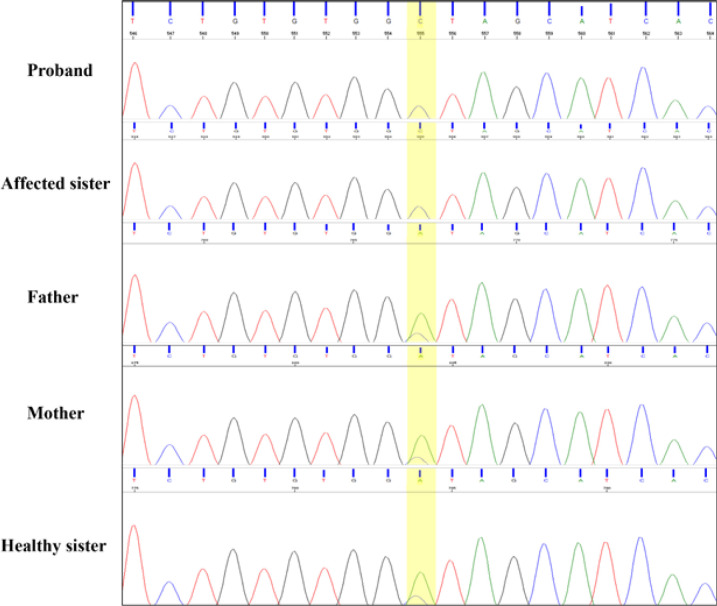
Materials & methods: The present study described two affected siblings from an Iranian family whose phenotypes overlap with intrauterine infections. They had almost similar presentations, including developmental delay, microcephaly, no fix and follow epileptic seizures and the same pattern of brain CT scan involvements. Following clinical and paraclinical assessments, whole-exome sequencing was employed to determine the disease-causing variant, and subsequently, PCR-Sanger sequencing was performed to indicate the segregation pattern of the candidate variant in family members.
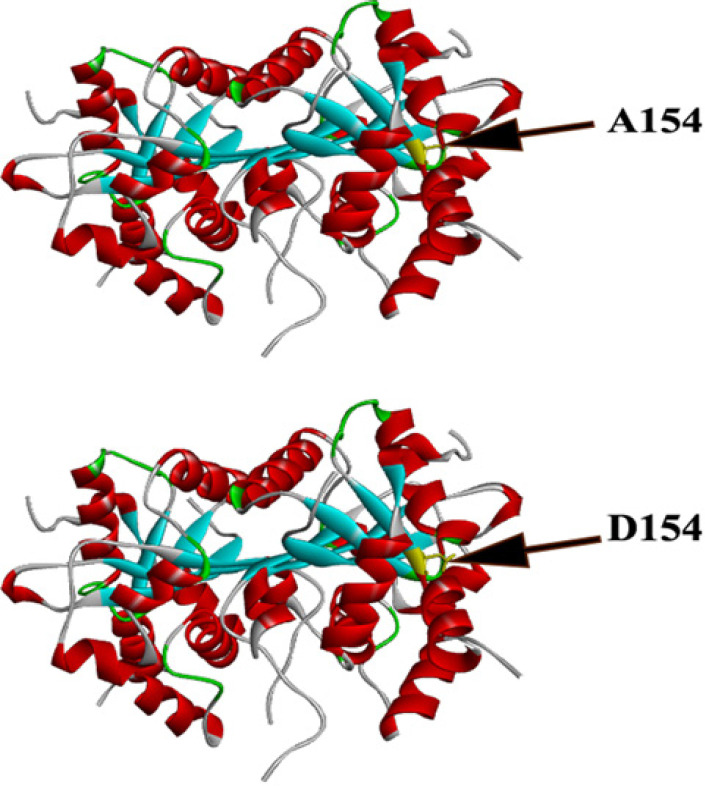
Results: Genetic analysis revealed a novel homozygous missense variant (c.461A>C; p.D154A) in the TREX1 gene in affected family members. Sanger sequencing of other family members showed the expected zygosities.
Conclusion: This study identifies a novel mutation in the TREX1 gene in this family and highlights the efficiency of next-generation sequencing-based techniques for obtaining a definite diagnosis in patients with early-onset encephalopathy.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


