{"title":"GM1和GM2-神经节苷脂病:临床特征、神经影像学发现和脑电图。","authors":"Parvaneh Karimzadeh, Masomeh Ebrahimi, Korosh Etemad, Farzad Ahmad Abadi, Zahra Hosseini Nezhad","doi":"10.22037/ijcn.v18i2.40751","DOIUrl":null,"url":null,"abstract":"<p><strong>Abstract: </strong>Gangliosidosis is one of the hereditary metabolic diseases caused by the accumulation of Gangliosid in the central nervous system, leading to severe and progressive neurological deficits. Regarding phenotype, GM1 and GM2-Gangliosidosis are divided into Infantile, Juvenile, and Adult.</p><p><strong>Materials & methods: </strong>In this study, thirty-seven patients with GM1 and GM2-Gangliosidosis were referred to the neurology department of Mofid Children's Hospital in Tehran, Iran, whose disease was confirmed from September 2019 to December 2021. This study assessed age, sex, and developmental status before the onset of the disease, clinical manifestations, brain imaging, and electroencephalography.</p><p><strong>Results: </strong>97.20% of patients were the result of family marriage. Approximately 80% of juvenile patients were developmentally normal before the onset of the disease. Developmental delay was more common among infantile GM1-Gangliosidosis than infantile GM2-Gangliosidosis, but in total, more than 50% of GM1&GM2-Gangliosidosis patients had reached their developmental milestone before the onset of the disease. With the onset of disease symptoms, 100% of patients regressed in terms of movement, 97.20% of them mentally, and 75% of them had seizures during the disease. The most common clinical findings were cherry-red spot, Mongolian spot, macrocephaly, organomegaly, hyperacusis, and scoliosis. The most common brain imaging findings included bilateral thalamus involvement, brain atrophy, PVL, and delayed myelination. The most common finding in electroencephalography was background low voltage with abnormal sharp waves.</p><p><strong>Conclusion: </strong>This study concluded that most of the patients are the result of family marriage, and most of the juvenile patients are developmentally normal before the onset of the disease. In addition, more than 50% of infantile patients reach their developmental milestones before the onset of the disease. The most common clinical findings of these patients are seizures, cherry-red spot, macrocephaly, hyperacusis, Mongolian spot, and bilateral involvement of the thalamus.</p>","PeriodicalId":14537,"journal":{"name":"Iranian Journal of Child Neurology","volume":"18 2","pages":"127-140"},"PeriodicalIF":0.9000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11015724/pdf/","citationCount":"0","resultStr":"{\"title\":\"GM1 and GM2-Gangliosidosis: Clinical Features, Neuroimaging Findings and Electroencephalography.\",\"authors\":\"Parvaneh Karimzadeh, Masomeh Ebrahimi, Korosh Etemad, Farzad Ahmad Abadi, Zahra Hosseini Nezhad\",\"doi\":\"10.22037/ijcn.v18i2.40751\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Abstract: </strong>Gangliosidosis is one of the hereditary metabolic diseases caused by the accumulation of Gangliosid in the central nervous system, leading to severe and progressive neurological deficits. Regarding phenotype, GM1 and GM2-Gangliosidosis are divided into Infantile, Juvenile, and Adult.</p><p><strong>Materials & methods: </strong>In this study, thirty-seven patients with GM1 and GM2-Gangliosidosis were referred to the neurology department of Mofid Children's Hospital in Tehran, Iran, whose disease was confirmed from September 2019 to December 2021. This study assessed age, sex, and developmental status before the onset of the disease, clinical manifestations, brain imaging, and electroencephalography.</p><p><strong>Results: </strong>97.20% of patients were the result of family marriage. Approximately 80% of juvenile patients were developmentally normal before the onset of the disease. Developmental delay was more common among infantile GM1-Gangliosidosis than infantile GM2-Gangliosidosis, but in total, more than 50% of GM1&GM2-Gangliosidosis patients had reached their developmental milestone before the onset of the disease. With the onset of disease symptoms, 100% of patients regressed in terms of movement, 97.20% of them mentally, and 75% of them had seizures during the disease. The most common clinical findings were cherry-red spot, Mongolian spot, macrocephaly, organomegaly, hyperacusis, and scoliosis. The most common brain imaging findings included bilateral thalamus involvement, brain atrophy, PVL, and delayed myelination. The most common finding in electroencephalography was background low voltage with abnormal sharp waves.</p><p><strong>Conclusion: </strong>This study concluded that most of the patients are the result of family marriage, and most of the juvenile patients are developmentally normal before the onset of the disease. In addition, more than 50% of infantile patients reach their developmental milestones before the onset of the disease. The most common clinical findings of these patients are seizures, cherry-red spot, macrocephaly, hyperacusis, Mongolian spot, and bilateral involvement of the thalamus.</p>\",\"PeriodicalId\":14537,\"journal\":{\"name\":\"Iranian Journal of Child Neurology\",\"volume\":\"18 2\",\"pages\":\"127-140\"},\"PeriodicalIF\":0.9000,\"publicationDate\":\"2024-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11015724/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Iranian Journal of Child Neurology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.22037/ijcn.v18i2.40751\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/3/12 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Iranian Journal of Child Neurology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.22037/ijcn.v18i2.40751","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/3/12 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
GM1 and GM2-Gangliosidosis: Clinical Features, Neuroimaging Findings and Electroencephalography.
Abstract: Gangliosidosis is one of the hereditary metabolic diseases caused by the accumulation of Gangliosid in the central nervous system, leading to severe and progressive neurological deficits. Regarding phenotype, GM1 and GM2-Gangliosidosis are divided into Infantile, Juvenile, and Adult.
Materials & methods: In this study, thirty-seven patients with GM1 and GM2-Gangliosidosis were referred to the neurology department of Mofid Children's Hospital in Tehran, Iran, whose disease was confirmed from September 2019 to December 2021. This study assessed age, sex, and developmental status before the onset of the disease, clinical manifestations, brain imaging, and electroencephalography.
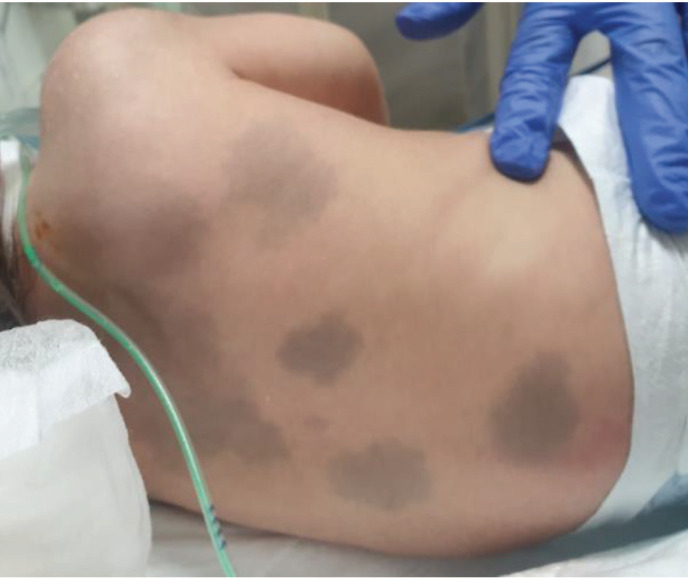
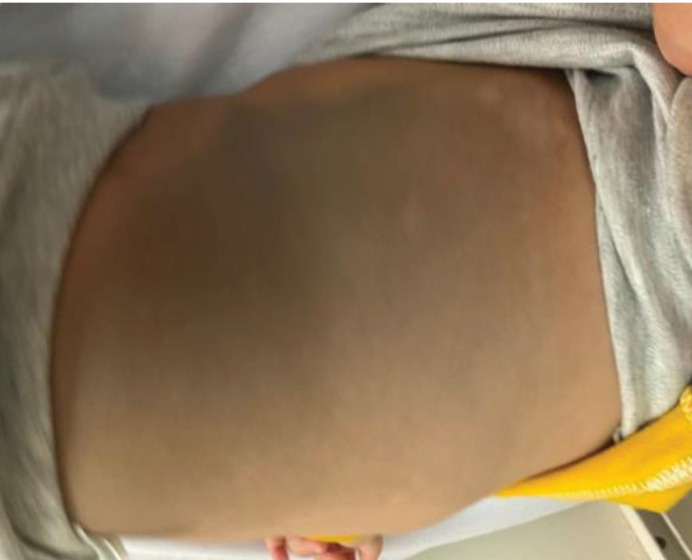
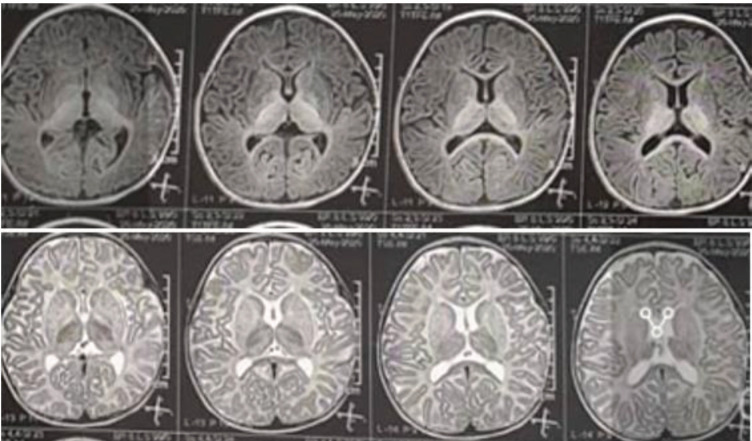
Results: 97.20% of patients were the result of family marriage. Approximately 80% of juvenile patients were developmentally normal before the onset of the disease. Developmental delay was more common among infantile GM1-Gangliosidosis than infantile GM2-Gangliosidosis, but in total, more than 50% of GM1&GM2-Gangliosidosis patients had reached their developmental milestone before the onset of the disease. With the onset of disease symptoms, 100% of patients regressed in terms of movement, 97.20% of them mentally, and 75% of them had seizures during the disease. The most common clinical findings were cherry-red spot, Mongolian spot, macrocephaly, organomegaly, hyperacusis, and scoliosis. The most common brain imaging findings included bilateral thalamus involvement, brain atrophy, PVL, and delayed myelination. The most common finding in electroencephalography was background low voltage with abnormal sharp waves.
Conclusion: This study concluded that most of the patients are the result of family marriage, and most of the juvenile patients are developmentally normal before the onset of the disease. In addition, more than 50% of infantile patients reach their developmental milestones before the onset of the disease. The most common clinical findings of these patients are seizures, cherry-red spot, macrocephaly, hyperacusis, Mongolian spot, and bilateral involvement of the thalamus.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


