Stephan Thaler, Felix Mayr, Siby Thomas, Alessio Gagliardi, Julija Zavadlav
{"title":"通过辍学蒙特卡洛,主动学习图神经网络用于金属有机框架的部分电荷预测","authors":"Stephan Thaler, Felix Mayr, Siby Thomas, Alessio Gagliardi, Julija Zavadlav","doi":"10.1038/s41524-024-01277-8","DOIUrl":null,"url":null,"abstract":"<p>Metal-organic frameworks (MOF) are an attractive class of porous materials due to their immense design space, allowing for application-tailored properties. Properties of interest, such as gas sorption, can be predicted in silico with molecular mechanics simulations. However, the accuracy is limited by the available empirical force field and partial charge estimation scheme. In this work, we train a graph neural network for partial charge prediction via active learning based on Dropout Monte Carlo. We show that active learning significantly reduces the required amount of labeled MOFs to reach a target accuracy. The obtained model generalizes well to different distributions of MOFs and Zeolites. In addition, the uncertainty predictions of Dropout Monte Carlo enable reliable estimation of the mean absolute error for unseen MOFs. This work paves the way towards accurate molecular modeling of MOFs via next-generation potentials with machine learning predicted partial charges, supporting in-silico material design.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"161 1","pages":""},"PeriodicalIF":9.4000,"publicationDate":"2024-05-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Active learning graph neural networks for partial charge prediction of metal-organic frameworks via dropout Monte Carlo\",\"authors\":\"Stephan Thaler, Felix Mayr, Siby Thomas, Alessio Gagliardi, Julija Zavadlav\",\"doi\":\"10.1038/s41524-024-01277-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Metal-organic frameworks (MOF) are an attractive class of porous materials due to their immense design space, allowing for application-tailored properties. Properties of interest, such as gas sorption, can be predicted in silico with molecular mechanics simulations. However, the accuracy is limited by the available empirical force field and partial charge estimation scheme. In this work, we train a graph neural network for partial charge prediction via active learning based on Dropout Monte Carlo. We show that active learning significantly reduces the required amount of labeled MOFs to reach a target accuracy. The obtained model generalizes well to different distributions of MOFs and Zeolites. In addition, the uncertainty predictions of Dropout Monte Carlo enable reliable estimation of the mean absolute error for unseen MOFs. This work paves the way towards accurate molecular modeling of MOFs via next-generation potentials with machine learning predicted partial charges, supporting in-silico material design.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"161 1\",\"pages\":\"\"},\"PeriodicalIF\":9.4000,\"publicationDate\":\"2024-05-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-024-01277-8\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01277-8","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
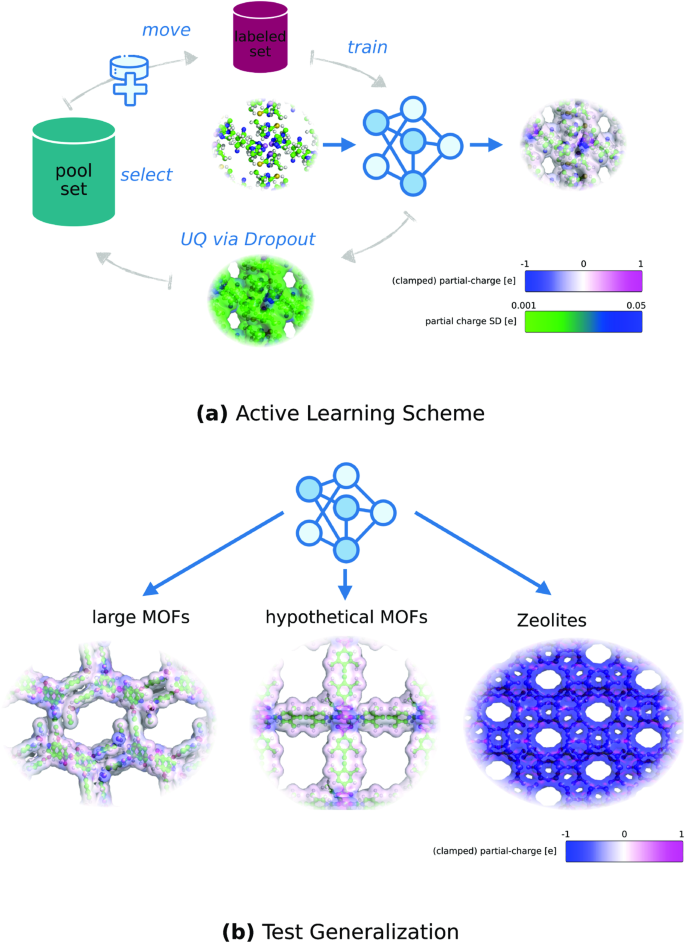
金属有机框架(MOF)是一类极具吸引力的多孔材料,因为它具有巨大的设计空间,可根据应用需要定制特性。气体吸附等相关特性可以通过分子力学模拟进行硅预测。然而,其准确性受到现有经验力场和部分电荷估算方案的限制。在这项工作中,我们通过基于 Dropout Monte Carlo 的主动学习训练图神经网络进行部分电荷预测。我们的研究表明,为达到目标精度,主动学习大大减少了所需的标记 MOFs 数量。所获得的模型对不同分布的 MOF 和沸石具有良好的通用性。此外,Dropout 蒙特卡罗的不确定性预测能够可靠地估算出未见 MOF 的平均绝对误差。这项工作为通过机器学习预测部分电荷的下一代电势建立精确的 MOFs 分子模型铺平了道路,从而支持硅内材料设计。
Active learning graph neural networks for partial charge prediction of metal-organic frameworks via dropout Monte Carlo
Metal-organic frameworks (MOF) are an attractive class of porous materials due to their immense design space, allowing for application-tailored properties. Properties of interest, such as gas sorption, can be predicted in silico with molecular mechanics simulations. However, the accuracy is limited by the available empirical force field and partial charge estimation scheme. In this work, we train a graph neural network for partial charge prediction via active learning based on Dropout Monte Carlo. We show that active learning significantly reduces the required amount of labeled MOFs to reach a target accuracy. The obtained model generalizes well to different distributions of MOFs and Zeolites. In addition, the uncertainty predictions of Dropout Monte Carlo enable reliable estimation of the mean absolute error for unseen MOFs. This work paves the way towards accurate molecular modeling of MOFs via next-generation potentials with machine learning predicted partial charges, supporting in-silico material design.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


