Mona A. Abdullaziz, Sana Takada, Boris Illarionov, Lais Pessanha de Carvalho, Yasumitsu Sakamoto, Stefan Höfmann, Talea Knak, Anna-Lene Kiffe-Delf, Flaminia Mazzone, Klaus Pfeffer, Rainer Kalscheuer, Adelbert Bacher, Jana Held, Markus Fischer, Nobutada Tanaka* and Thomas Kurz*,
{"title":"针对 1-Deoxy-d-xylulose 5-Phosphate Reductoisomerase (DXR) 先前未知子口的反向 N-取代羟肟酸衍生物。","authors":"Mona A. Abdullaziz, Sana Takada, Boris Illarionov, Lais Pessanha de Carvalho, Yasumitsu Sakamoto, Stefan Höfmann, Talea Knak, Anna-Lene Kiffe-Delf, Flaminia Mazzone, Klaus Pfeffer, Rainer Kalscheuer, Adelbert Bacher, Jana Held, Markus Fischer, Nobutada Tanaka* and Thomas Kurz*, ","doi":"10.1021/acsinfecdis.4c00100","DOIUrl":null,"url":null,"abstract":"<p >Reverse analogs of the phosphonohydroxamic acid antibiotic fosmidomycin are potent inhibitors of the nonmevalonate isoprenoid biosynthesis enzyme 1-deoxy-<span>d-</span>xylulose 5-phosphate reductoisomerase (DXR, IspC) of <i>Plasmodium falciparum</i>. Some novel analogs with large phenylalkyl substituents at the hydroxamic acid nitrogen exhibit nanomolar <i>Pf</i>DXR inhibition and potent <i>in vitro</i> growth inhibition of <i>P. falciparum</i> parasites coupled with good parasite selectivity. X-ray crystallographic studies demonstrated that the <i>N</i>-phenylpropyl substituent of the newly developed lead compound <b>13e</b> is accommodated in a subpocket within the DXR catalytic domain but does not reach the NADPH binding pocket of the <i>N</i>-terminal domain. As shown for reverse carba and thia analogs, <i>Pf</i>DXR selectively binds the <i>S</i>-enantiomer of the new lead compound. In addition, some representatives of the novel inhibitor subclass are nanomolar <i>Escherichia coli</i> DXR inhibitors, whereas the inhibition of <i>Mycobacterium tuberculosis</i> DXR is considerably weaker.</p>","PeriodicalId":17,"journal":{"name":"ACS Infectious Diseases","volume":"10 5","pages":"1739–1752"},"PeriodicalIF":3.8000,"publicationDate":"2024-04-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Reverse N-Substituted Hydroxamic Acid Derivatives of Fosmidomycin Target a Previously Unknown Subpocket of 1-Deoxy-d-xylulose 5-Phosphate Reductoisomerase (DXR)\",\"authors\":\"Mona A. Abdullaziz, Sana Takada, Boris Illarionov, Lais Pessanha de Carvalho, Yasumitsu Sakamoto, Stefan Höfmann, Talea Knak, Anna-Lene Kiffe-Delf, Flaminia Mazzone, Klaus Pfeffer, Rainer Kalscheuer, Adelbert Bacher, Jana Held, Markus Fischer, Nobutada Tanaka* and Thomas Kurz*, \",\"doi\":\"10.1021/acsinfecdis.4c00100\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Reverse analogs of the phosphonohydroxamic acid antibiotic fosmidomycin are potent inhibitors of the nonmevalonate isoprenoid biosynthesis enzyme 1-deoxy-<span>d-</span>xylulose 5-phosphate reductoisomerase (DXR, IspC) of <i>Plasmodium falciparum</i>. Some novel analogs with large phenylalkyl substituents at the hydroxamic acid nitrogen exhibit nanomolar <i>Pf</i>DXR inhibition and potent <i>in vitro</i> growth inhibition of <i>P. falciparum</i> parasites coupled with good parasite selectivity. X-ray crystallographic studies demonstrated that the <i>N</i>-phenylpropyl substituent of the newly developed lead compound <b>13e</b> is accommodated in a subpocket within the DXR catalytic domain but does not reach the NADPH binding pocket of the <i>N</i>-terminal domain. As shown for reverse carba and thia analogs, <i>Pf</i>DXR selectively binds the <i>S</i>-enantiomer of the new lead compound. In addition, some representatives of the novel inhibitor subclass are nanomolar <i>Escherichia coli</i> DXR inhibitors, whereas the inhibition of <i>Mycobacterium tuberculosis</i> DXR is considerably weaker.</p>\",\"PeriodicalId\":17,\"journal\":{\"name\":\"ACS Infectious Diseases\",\"volume\":\"10 5\",\"pages\":\"1739–1752\"},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2024-04-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Infectious Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acsinfecdis.4c00100\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Infectious Diseases","FirstCategoryId":"3","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acsinfecdis.4c00100","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
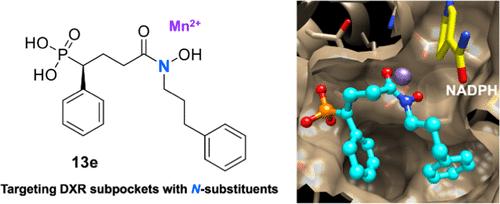
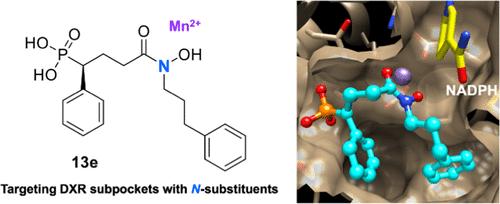
Reverse N-Substituted Hydroxamic Acid Derivatives of Fosmidomycin Target a Previously Unknown Subpocket of 1-Deoxy-d-xylulose 5-Phosphate Reductoisomerase (DXR)
Reverse analogs of the phosphonohydroxamic acid antibiotic fosmidomycin are potent inhibitors of the nonmevalonate isoprenoid biosynthesis enzyme 1-deoxy-d-xylulose 5-phosphate reductoisomerase (DXR, IspC) of Plasmodium falciparum. Some novel analogs with large phenylalkyl substituents at the hydroxamic acid nitrogen exhibit nanomolar PfDXR inhibition and potent in vitro growth inhibition of P. falciparum parasites coupled with good parasite selectivity. X-ray crystallographic studies demonstrated that the N-phenylpropyl substituent of the newly developed lead compound 13e is accommodated in a subpocket within the DXR catalytic domain but does not reach the NADPH binding pocket of the N-terminal domain. As shown for reverse carba and thia analogs, PfDXR selectively binds the S-enantiomer of the new lead compound. In addition, some representatives of the novel inhibitor subclass are nanomolar Escherichia coli DXR inhibitors, whereas the inhibition of Mycobacterium tuberculosis DXR is considerably weaker.
期刊介绍:
ACS Infectious Diseases will be the first journal to highlight chemistry and its role in this multidisciplinary and collaborative research area. The journal will cover a diverse array of topics including, but not limited to:
* Discovery and development of new antimicrobial agents — identified through target- or phenotypic-based approaches as well as compounds that induce synergy with antimicrobials.
* Characterization and validation of drug target or pathways — use of single target and genome-wide knockdown and knockouts, biochemical studies, structural biology, new technologies to facilitate characterization and prioritization of potential drug targets.
* Mechanism of drug resistance — fundamental research that advances our understanding of resistance; strategies to prevent resistance.
* Mechanisms of action — use of genetic, metabolomic, and activity- and affinity-based protein profiling to elucidate the mechanism of action of clinical and experimental antimicrobial agents.
* Host-pathogen interactions — tools for studying host-pathogen interactions, cellular biochemistry of hosts and pathogens, and molecular interactions of pathogens with host microbiota.
* Small molecule vaccine adjuvants for infectious disease.
* Viral and bacterial biochemistry and molecular biology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


