Jeremy Jones, Robert D. Clark, Michael S. Lawless, David W. Miller, Marvin Waldman
{"title":"人工智能驱动的药物设计(AIDD)平台:一个交互式多参数优化系统,将分子进化与基于生理学的药代动力学模拟融为一体。","authors":"Jeremy Jones, Robert D. Clark, Michael S. Lawless, David W. Miller, Marvin Waldman","doi":"10.1007/s10822-024-00552-6","DOIUrl":null,"url":null,"abstract":"<div><p>Computer-aided drug design has advanced rapidly in recent years, and multiple instances of in silico designed molecules advancing to the clinic have demonstrated the contribution of this field to medicine. Properly designed and implemented platforms can drastically reduce drug development timelines and costs. While such efforts were initially focused primarily on target affinity/activity, it is now appreciated that other parameters are equally important in the successful development of a drug and its progression to the clinic, including pharmacokinetic properties as well as absorption, distribution, metabolic, excretion and toxicological (ADMET) properties. In the last decade, several programs have been developed that incorporate these properties into the drug design and optimization process and to varying degrees, allowing for multi-parameter optimization. Here, we introduce the Artificial Intelligence-driven Drug Design (AIDD) platform, which automates the drug design process by integrating high-throughput physiologically-based pharmacokinetic simulations (powered by GastroPlus) and ADMET predictions (powered by ADMET Predictor) with an advanced evolutionary algorithm that is quite different than current generative models. AIDD uses these and other estimates in iteratively performing multi-objective optimizations to produce novel molecules that are active and lead-like. Here we describe the AIDD workflow and details of the methodologies involved therein. We use a dataset of triazolopyrimidine inhibitors of the dihydroorotate dehydrogenase from <i>Plasmodium falciparum</i> to illustrate how AIDD generates novel sets of molecules.</p></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"38 1","pages":""},"PeriodicalIF":3.1000,"publicationDate":"2024-03-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"The AI-driven Drug Design (AIDD) platform: an interactive multi-parameter optimization system integrating molecular evolution with physiologically based pharmacokinetic simulations\",\"authors\":\"Jeremy Jones, Robert D. Clark, Michael S. Lawless, David W. Miller, Marvin Waldman\",\"doi\":\"10.1007/s10822-024-00552-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Computer-aided drug design has advanced rapidly in recent years, and multiple instances of in silico designed molecules advancing to the clinic have demonstrated the contribution of this field to medicine. Properly designed and implemented platforms can drastically reduce drug development timelines and costs. While such efforts were initially focused primarily on target affinity/activity, it is now appreciated that other parameters are equally important in the successful development of a drug and its progression to the clinic, including pharmacokinetic properties as well as absorption, distribution, metabolic, excretion and toxicological (ADMET) properties. In the last decade, several programs have been developed that incorporate these properties into the drug design and optimization process and to varying degrees, allowing for multi-parameter optimization. Here, we introduce the Artificial Intelligence-driven Drug Design (AIDD) platform, which automates the drug design process by integrating high-throughput physiologically-based pharmacokinetic simulations (powered by GastroPlus) and ADMET predictions (powered by ADMET Predictor) with an advanced evolutionary algorithm that is quite different than current generative models. AIDD uses these and other estimates in iteratively performing multi-objective optimizations to produce novel molecules that are active and lead-like. Here we describe the AIDD workflow and details of the methodologies involved therein. We use a dataset of triazolopyrimidine inhibitors of the dihydroorotate dehydrogenase from <i>Plasmodium falciparum</i> to illustrate how AIDD generates novel sets of molecules.</p></div>\",\"PeriodicalId\":621,\"journal\":{\"name\":\"Journal of Computer-Aided Molecular Design\",\"volume\":\"38 1\",\"pages\":\"\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-03-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computer-Aided Molecular Design\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10822-024-00552-6\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-024-00552-6","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
The AI-driven Drug Design (AIDD) platform: an interactive multi-parameter optimization system integrating molecular evolution with physiologically based pharmacokinetic simulations
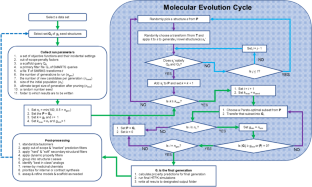
Computer-aided drug design has advanced rapidly in recent years, and multiple instances of in silico designed molecules advancing to the clinic have demonstrated the contribution of this field to medicine. Properly designed and implemented platforms can drastically reduce drug development timelines and costs. While such efforts were initially focused primarily on target affinity/activity, it is now appreciated that other parameters are equally important in the successful development of a drug and its progression to the clinic, including pharmacokinetic properties as well as absorption, distribution, metabolic, excretion and toxicological (ADMET) properties. In the last decade, several programs have been developed that incorporate these properties into the drug design and optimization process and to varying degrees, allowing for multi-parameter optimization. Here, we introduce the Artificial Intelligence-driven Drug Design (AIDD) platform, which automates the drug design process by integrating high-throughput physiologically-based pharmacokinetic simulations (powered by GastroPlus) and ADMET predictions (powered by ADMET Predictor) with an advanced evolutionary algorithm that is quite different than current generative models. AIDD uses these and other estimates in iteratively performing multi-objective optimizations to produce novel molecules that are active and lead-like. Here we describe the AIDD workflow and details of the methodologies involved therein. We use a dataset of triazolopyrimidine inhibitors of the dihydroorotate dehydrogenase from Plasmodium falciparum to illustrate how AIDD generates novel sets of molecules.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


