{"title":"一名反复发作的顽固性呕吐伊朗患者缺乏脱氢酶 (DLD):通过补充硫胺素成功治疗。","authors":"Toktam Moosavian, Ghazaleh Jamalipour Soufi, Sharareh Kamfar","doi":"10.22037/ijcn.v18i1.38971","DOIUrl":null,"url":null,"abstract":"<p><p>Dihydrolipoamide dehydrogenase (DLD) deficiency is a rare disease of genetic origin due to the malfunctioning of a shared subunit of three mitochondrial multi-enzyme complexes. Phenotypes of this disease are a set of clinical manifestations ranging from neonatal disorders to myopathy or recurrent episodes of liver failures, and vomiting for which no adequate or definitive treatment is currently available. This study described a case involving a 16-year-old boy who had experienced recurrent vomiting of unknown cause from age two. Normal value ranges for the basic metabolic panel were reported in previous years. The patient was admitted with Wernicke's encephalopathy after the last vomiting attack, also indicating metabolites of organic acids compatible with DLD deficiency. Whole exome sequencing identified a known pathogenic mutation in the DLD gene, leading to a diagnosis of DLD deficiency. Our patient was treated with a high dose of thiamine supplementation and continued treatment, has not experienced any vomiting attacks or related problems in the last two years and has adequately responded to the treatment prescribed. Normal urine organic acid levels in patients with recurrent vomiting cannot roll out DLD deficiency. However, although thiamine deficiency typically induces Wernicke's encephalopathy, it can also be implicated in pyruvate dehydrogenase complex (PDHc) deficiency, and high-dose thiamine therapy (with doses up to 30 mg/kg) is recommended for deficient patients.</p>","PeriodicalId":14537,"journal":{"name":"Iranian Journal of Child Neurology","volume":"18 1","pages":"131-138"},"PeriodicalIF":0.9000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10874511/pdf/","citationCount":"0","resultStr":"{\"title\":\"Dehydrogenase (DLD) Deficiency in an Iranian Patient with Recurrent Intractable Vomiting: Successful Treatment with Thiamine Supplementation.\",\"authors\":\"Toktam Moosavian, Ghazaleh Jamalipour Soufi, Sharareh Kamfar\",\"doi\":\"10.22037/ijcn.v18i1.38971\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Dihydrolipoamide dehydrogenase (DLD) deficiency is a rare disease of genetic origin due to the malfunctioning of a shared subunit of three mitochondrial multi-enzyme complexes. Phenotypes of this disease are a set of clinical manifestations ranging from neonatal disorders to myopathy or recurrent episodes of liver failures, and vomiting for which no adequate or definitive treatment is currently available. This study described a case involving a 16-year-old boy who had experienced recurrent vomiting of unknown cause from age two. Normal value ranges for the basic metabolic panel were reported in previous years. The patient was admitted with Wernicke's encephalopathy after the last vomiting attack, also indicating metabolites of organic acids compatible with DLD deficiency. Whole exome sequencing identified a known pathogenic mutation in the DLD gene, leading to a diagnosis of DLD deficiency. Our patient was treated with a high dose of thiamine supplementation and continued treatment, has not experienced any vomiting attacks or related problems in the last two years and has adequately responded to the treatment prescribed. Normal urine organic acid levels in patients with recurrent vomiting cannot roll out DLD deficiency. However, although thiamine deficiency typically induces Wernicke's encephalopathy, it can also be implicated in pyruvate dehydrogenase complex (PDHc) deficiency, and high-dose thiamine therapy (with doses up to 30 mg/kg) is recommended for deficient patients.</p>\",\"PeriodicalId\":14537,\"journal\":{\"name\":\"Iranian Journal of Child Neurology\",\"volume\":\"18 1\",\"pages\":\"131-138\"},\"PeriodicalIF\":0.9000,\"publicationDate\":\"2024-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10874511/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Iranian Journal of Child Neurology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.22037/ijcn.v18i1.38971\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/18 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Iranian Journal of Child Neurology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.22037/ijcn.v18i1.38971","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/18 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Dehydrogenase (DLD) Deficiency in an Iranian Patient with Recurrent Intractable Vomiting: Successful Treatment with Thiamine Supplementation.
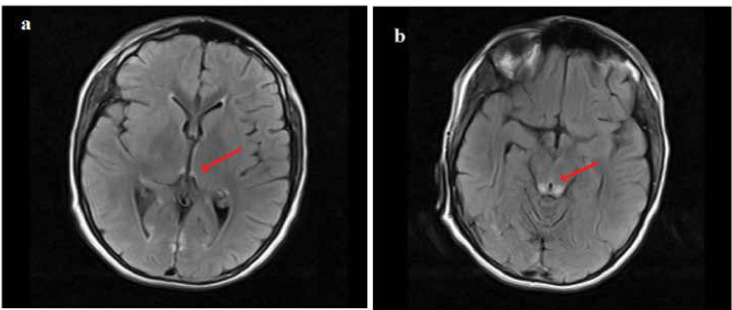
Dihydrolipoamide dehydrogenase (DLD) deficiency is a rare disease of genetic origin due to the malfunctioning of a shared subunit of three mitochondrial multi-enzyme complexes. Phenotypes of this disease are a set of clinical manifestations ranging from neonatal disorders to myopathy or recurrent episodes of liver failures, and vomiting for which no adequate or definitive treatment is currently available. This study described a case involving a 16-year-old boy who had experienced recurrent vomiting of unknown cause from age two. Normal value ranges for the basic metabolic panel were reported in previous years. The patient was admitted with Wernicke's encephalopathy after the last vomiting attack, also indicating metabolites of organic acids compatible with DLD deficiency. Whole exome sequencing identified a known pathogenic mutation in the DLD gene, leading to a diagnosis of DLD deficiency. Our patient was treated with a high dose of thiamine supplementation and continued treatment, has not experienced any vomiting attacks or related problems in the last two years and has adequately responded to the treatment prescribed. Normal urine organic acid levels in patients with recurrent vomiting cannot roll out DLD deficiency. However, although thiamine deficiency typically induces Wernicke's encephalopathy, it can also be implicated in pyruvate dehydrogenase complex (PDHc) deficiency, and high-dose thiamine therapy (with doses up to 30 mg/kg) is recommended for deficient patients.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


