{"title":"蒽的分子对接和 DFT 计算:量子化学方法的启示","authors":"Rajendran Vijayakumar, Arangarajan Viji, Karuppaiya Vanasundari, Vadivel Balachandran, Antony Prabhu Arockia Dass","doi":"10.1002/crat.202300150","DOIUrl":null,"url":null,"abstract":"<p>The molecular structure and spectroscopic data of (2E)-1-(Anthracen-9-yl)-3-(4-nitrophenyl)prop-2-en-1-one are obtained from DFT (B3LYP) with 6-31G(d,p) and 6-31G+(d,p) basis set calculations. The geometry of the molecule is fully optimized, vibrational spectra are calculated and fundamental vibrations are assigned on the basis of potential energy distribution (PED) of the vibrational modes. Molecular parameters such as bond length and bond angle are calculated with the same level of theory. The intramolecular charge transfer is calculated by means of natural bond orbital analysis (NBO). Besides, the molecular electrostatic potential (MEP), HOMO - LUMO, Fukui functions, RDG and ELF are performed. The biological effect is made on the basis of prediction of molecular docking results.</p>","PeriodicalId":48935,"journal":{"name":"Crystal Research and Technology","volume":"59 5","pages":""},"PeriodicalIF":1.5000,"publicationDate":"2023-12-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Molecular Docking and DFT Calculations of Anthracene: Insights from Quantum Chemical Methods\",\"authors\":\"Rajendran Vijayakumar, Arangarajan Viji, Karuppaiya Vanasundari, Vadivel Balachandran, Antony Prabhu Arockia Dass\",\"doi\":\"10.1002/crat.202300150\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The molecular structure and spectroscopic data of (2E)-1-(Anthracen-9-yl)-3-(4-nitrophenyl)prop-2-en-1-one are obtained from DFT (B3LYP) with 6-31G(d,p) and 6-31G+(d,p) basis set calculations. The geometry of the molecule is fully optimized, vibrational spectra are calculated and fundamental vibrations are assigned on the basis of potential energy distribution (PED) of the vibrational modes. Molecular parameters such as bond length and bond angle are calculated with the same level of theory. The intramolecular charge transfer is calculated by means of natural bond orbital analysis (NBO). Besides, the molecular electrostatic potential (MEP), HOMO - LUMO, Fukui functions, RDG and ELF are performed. The biological effect is made on the basis of prediction of molecular docking results.</p>\",\"PeriodicalId\":48935,\"journal\":{\"name\":\"Crystal Research and Technology\",\"volume\":\"59 5\",\"pages\":\"\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2023-12-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Crystal Research and Technology\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/crat.202300150\",\"RegionNum\":4,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"Chemistry\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Crystal Research and Technology","FirstCategoryId":"88","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/crat.202300150","RegionNum":4,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Chemistry","Score":null,"Total":0}
Molecular Docking and DFT Calculations of Anthracene: Insights from Quantum Chemical Methods
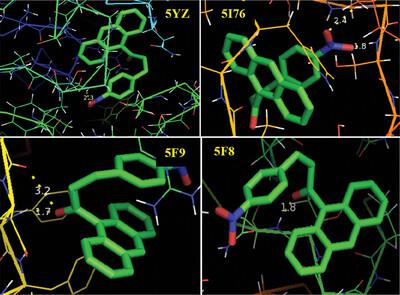
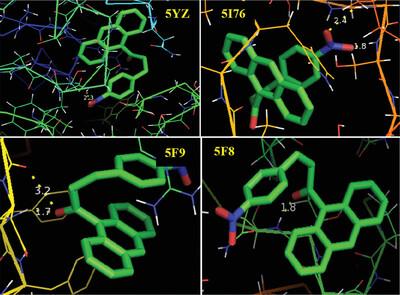
The molecular structure and spectroscopic data of (2E)-1-(Anthracen-9-yl)-3-(4-nitrophenyl)prop-2-en-1-one are obtained from DFT (B3LYP) with 6-31G(d,p) and 6-31G+(d,p) basis set calculations. The geometry of the molecule is fully optimized, vibrational spectra are calculated and fundamental vibrations are assigned on the basis of potential energy distribution (PED) of the vibrational modes. Molecular parameters such as bond length and bond angle are calculated with the same level of theory. The intramolecular charge transfer is calculated by means of natural bond orbital analysis (NBO). Besides, the molecular electrostatic potential (MEP), HOMO - LUMO, Fukui functions, RDG and ELF are performed. The biological effect is made on the basis of prediction of molecular docking results.
期刊介绍:
The journal Crystal Research and Technology is a pure online Journal (since 2012).
Crystal Research and Technology is an international journal examining all aspects of research within experimental, industrial, and theoretical crystallography. The journal covers the relevant aspects of
-crystal growth techniques and phenomena (including bulk growth, thin films)
-modern crystalline materials (e.g. smart materials, nanocrystals, quasicrystals, liquid crystals)
-industrial crystallisation
-application of crystals in materials science, electronics, data storage, and optics
-experimental, simulation and theoretical studies of the structural properties of crystals
-crystallographic computing

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


