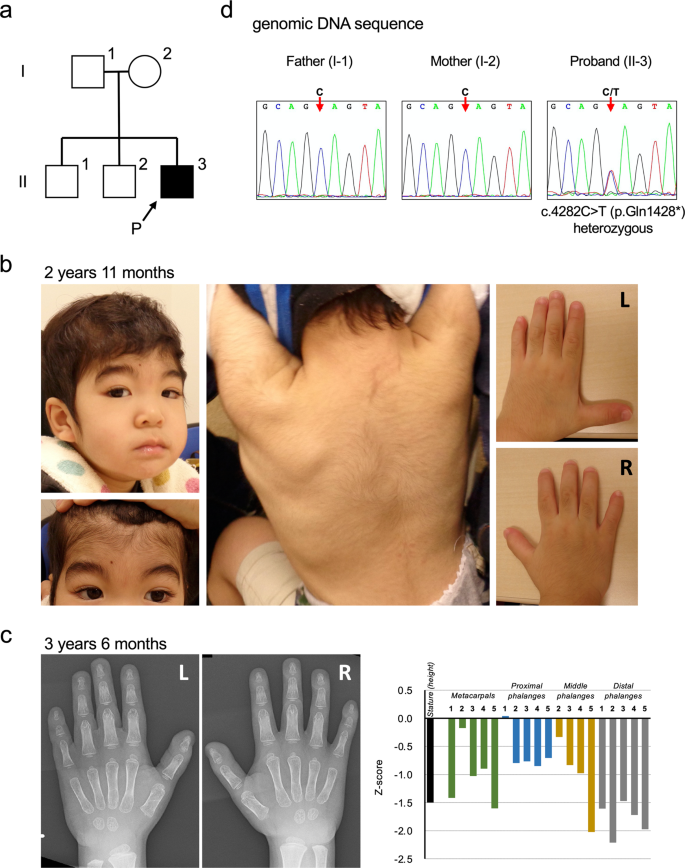
{"title":"ARID1B中一种新的无义变异引起同时的RNA衰变和外显子跳变与Coffin-Siris综合征有关。","authors":"Viktoriia Sofronova, Yu Fukushima, Mitsuo Masuno, Mami Naka, Miho Nagata, Yasuki Ishihara, Yohei Miyashita, Yoshihiro Asano, Takahito Moriwaki, Rina Iwata, Seigo Terawaki, Yasuko Yamanouchi, Takanobu Otomo","doi":"10.1038/s41439-022-00203-y","DOIUrl":null,"url":null,"abstract":"<p><p>Coffin-Siris syndrome (CSS) is a congenital disorder that is characterized by an absent/hypoplastic fifth distal phalanx, psychomotor developmental delay, and coarse facial features. One of the causative genes, ARID1B (AT-rich interactive domain-containing protein 1B), encodes components of the BAF chromatin remodeling complexes. Here, we report a case of a 3-year 8-month-old male with a novel nonsense variant (NM_001374820.1:c.4282C > T, p.(Gln1428*)) in the ARID1B gene, which was identified with whole-exome sequencing. He showed clinical symptoms of cleft soft palate, distinctive facial features (flat nasal bridge, thick eyebrows, and long eyelashes), right cryptorchidism, and hypertrichosis that partially overlapped with CSS. One of the most characteristic features of CSS is absent/hypoplastic fifth distal phalanx. He showed no obvious clinical finding in the lengths of his fingers or in the formation of his fingernails. However, radiographic analyses of the metacarpophalangeal bones revealed shortening of all the distal phalanges and fifth middle phalanges, suggesting brachydactyly. We performed mRNA analyses and revealed that both nonsense-mediated decay and nonsense-associated altered splicing were simultaneously caused by the c.4282C > T nonsense variant. The proband's clinical manifestations fit the previously reported criteria of disease for CSS or intellectual disability with ARID1B variant. Altogether, we suggest that c.4282C > T is a pathogenic variant that causes this clinical phenotype.</p>","PeriodicalId":36861,"journal":{"name":"Human Genome Variation","volume":null,"pages":null},"PeriodicalIF":1.0000,"publicationDate":"2022-07-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9314373/pdf/","citationCount":"1","resultStr":"{\"title\":\"A novel nonsense variant in ARID1B causing simultaneous RNA decay and exon skipping is associated with Coffin-Siris syndrome.\",\"authors\":\"Viktoriia Sofronova, Yu Fukushima, Mitsuo Masuno, Mami Naka, Miho Nagata, Yasuki Ishihara, Yohei Miyashita, Yoshihiro Asano, Takahito Moriwaki, Rina Iwata, Seigo Terawaki, Yasuko Yamanouchi, Takanobu Otomo\",\"doi\":\"10.1038/s41439-022-00203-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Coffin-Siris syndrome (CSS) is a congenital disorder that is characterized by an absent/hypoplastic fifth distal phalanx, psychomotor developmental delay, and coarse facial features. One of the causative genes, ARID1B (AT-rich interactive domain-containing protein 1B), encodes components of the BAF chromatin remodeling complexes. Here, we report a case of a 3-year 8-month-old male with a novel nonsense variant (NM_001374820.1:c.4282C > T, p.(Gln1428*)) in the ARID1B gene, which was identified with whole-exome sequencing. He showed clinical symptoms of cleft soft palate, distinctive facial features (flat nasal bridge, thick eyebrows, and long eyelashes), right cryptorchidism, and hypertrichosis that partially overlapped with CSS. One of the most characteristic features of CSS is absent/hypoplastic fifth distal phalanx. He showed no obvious clinical finding in the lengths of his fingers or in the formation of his fingernails. However, radiographic analyses of the metacarpophalangeal bones revealed shortening of all the distal phalanges and fifth middle phalanges, suggesting brachydactyly. We performed mRNA analyses and revealed that both nonsense-mediated decay and nonsense-associated altered splicing were simultaneously caused by the c.4282C > T nonsense variant. The proband's clinical manifestations fit the previously reported criteria of disease for CSS or intellectual disability with ARID1B variant. Altogether, we suggest that c.4282C > T is a pathogenic variant that causes this clinical phenotype.</p>\",\"PeriodicalId\":36861,\"journal\":{\"name\":\"Human Genome Variation\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":1.0000,\"publicationDate\":\"2022-07-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9314373/pdf/\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Genome Variation\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1038/s41439-022-00203-y\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genome Variation","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1038/s41439-022-00203-y","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
A novel nonsense variant in ARID1B causing simultaneous RNA decay and exon skipping is associated with Coffin-Siris syndrome.
Coffin-Siris syndrome (CSS) is a congenital disorder that is characterized by an absent/hypoplastic fifth distal phalanx, psychomotor developmental delay, and coarse facial features. One of the causative genes, ARID1B (AT-rich interactive domain-containing protein 1B), encodes components of the BAF chromatin remodeling complexes. Here, we report a case of a 3-year 8-month-old male with a novel nonsense variant (NM_001374820.1:c.4282C > T, p.(Gln1428*)) in the ARID1B gene, which was identified with whole-exome sequencing. He showed clinical symptoms of cleft soft palate, distinctive facial features (flat nasal bridge, thick eyebrows, and long eyelashes), right cryptorchidism, and hypertrichosis that partially overlapped with CSS. One of the most characteristic features of CSS is absent/hypoplastic fifth distal phalanx. He showed no obvious clinical finding in the lengths of his fingers or in the formation of his fingernails. However, radiographic analyses of the metacarpophalangeal bones revealed shortening of all the distal phalanges and fifth middle phalanges, suggesting brachydactyly. We performed mRNA analyses and revealed that both nonsense-mediated decay and nonsense-associated altered splicing were simultaneously caused by the c.4282C > T nonsense variant. The proband's clinical manifestations fit the previously reported criteria of disease for CSS or intellectual disability with ARID1B variant. Altogether, we suggest that c.4282C > T is a pathogenic variant that causes this clinical phenotype.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


