Qianqian Zhang, Jianting Han, Yongchang Zhu, Fansen Yu, Xiaopeng Hu, Henry H. Y. Tong, Huanxiang Liu
{"title":"通过基于集成对接的虚拟筛选和生物测定发现新的强效InhA直接抑制剂。","authors":"Qianqian Zhang, Jianting Han, Yongchang Zhu, Fansen Yu, Xiaopeng Hu, Henry H. Y. Tong, Huanxiang Liu","doi":"10.1007/s10822-023-00530-4","DOIUrl":null,"url":null,"abstract":"<div><p>Multidrug-resistant tuberculosis (MDR-TB) continues to spread worldwide and remains one of the leading causes of death among infectious diseases. The enoyl-acyl carrier protein reductase (InhA) belongs to FAS-II family and is essential for the formation of the Mycobacterium tuberculosis cell wall. Recent years, InhA direct inhibitors have been extensively studied to overcome MDR-TB. However, there are still no inhibitors that have entered clinical research. Here, the ensemble docking-based virtual screening along with biological assay were used to identify potent InhA direct inhibitors from Chembridge, Chemdiv, and Specs. Ultimately, 34 compounds were purchased and first assayed for the binding affinity, of which four compounds can bind InhA well with K<sub>D</sub> values ranging from 48.4 to 56.2 µM. Among them, compound 9,222,034 has the best inhibitory activity against InhA enzyme with an IC<sub>50</sub> value of 18.05 µM. In addition, the molecular dynamic simulation and binding free energy calculation indicate that the identified compounds bind to InhA with “extended” conformation. Residue energy decomposition shows that residues such as Tyr158, Met161, and Met191 have higher energy contributions in the binding of compounds. By analyzing the binding modes, we found that these compounds can bind to a hydrophobic sub-pocket formed by residues Tyr158, Phe149, Ile215, Leu218, etc., resulting in extensive van der Waals interactions. In summary, this study proposed an efficient strategy for discovering InhA direct inhibitors through ensemble docking-based virtual screening, and finally identified four active compounds with new skeletons, which can provide valuable information for the discovery and optimization of InhA direct inhibitors.</p></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"37 12","pages":"695 - 706"},"PeriodicalIF":3.0000,"publicationDate":"2023-08-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Discovery of novel and potent InhA direct inhibitors by ensemble docking-based virtual screening and biological assays\",\"authors\":\"Qianqian Zhang, Jianting Han, Yongchang Zhu, Fansen Yu, Xiaopeng Hu, Henry H. Y. Tong, Huanxiang Liu\",\"doi\":\"10.1007/s10822-023-00530-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Multidrug-resistant tuberculosis (MDR-TB) continues to spread worldwide and remains one of the leading causes of death among infectious diseases. The enoyl-acyl carrier protein reductase (InhA) belongs to FAS-II family and is essential for the formation of the Mycobacterium tuberculosis cell wall. Recent years, InhA direct inhibitors have been extensively studied to overcome MDR-TB. However, there are still no inhibitors that have entered clinical research. Here, the ensemble docking-based virtual screening along with biological assay were used to identify potent InhA direct inhibitors from Chembridge, Chemdiv, and Specs. Ultimately, 34 compounds were purchased and first assayed for the binding affinity, of which four compounds can bind InhA well with K<sub>D</sub> values ranging from 48.4 to 56.2 µM. Among them, compound 9,222,034 has the best inhibitory activity against InhA enzyme with an IC<sub>50</sub> value of 18.05 µM. In addition, the molecular dynamic simulation and binding free energy calculation indicate that the identified compounds bind to InhA with “extended” conformation. Residue energy decomposition shows that residues such as Tyr158, Met161, and Met191 have higher energy contributions in the binding of compounds. By analyzing the binding modes, we found that these compounds can bind to a hydrophobic sub-pocket formed by residues Tyr158, Phe149, Ile215, Leu218, etc., resulting in extensive van der Waals interactions. In summary, this study proposed an efficient strategy for discovering InhA direct inhibitors through ensemble docking-based virtual screening, and finally identified four active compounds with new skeletons, which can provide valuable information for the discovery and optimization of InhA direct inhibitors.</p></div>\",\"PeriodicalId\":621,\"journal\":{\"name\":\"Journal of Computer-Aided Molecular Design\",\"volume\":\"37 12\",\"pages\":\"695 - 706\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2023-08-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computer-Aided Molecular Design\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10822-023-00530-4\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-023-00530-4","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Discovery of novel and potent InhA direct inhibitors by ensemble docking-based virtual screening and biological assays
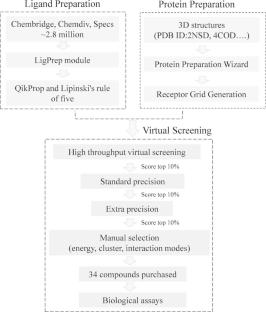
Multidrug-resistant tuberculosis (MDR-TB) continues to spread worldwide and remains one of the leading causes of death among infectious diseases. The enoyl-acyl carrier protein reductase (InhA) belongs to FAS-II family and is essential for the formation of the Mycobacterium tuberculosis cell wall. Recent years, InhA direct inhibitors have been extensively studied to overcome MDR-TB. However, there are still no inhibitors that have entered clinical research. Here, the ensemble docking-based virtual screening along with biological assay were used to identify potent InhA direct inhibitors from Chembridge, Chemdiv, and Specs. Ultimately, 34 compounds were purchased and first assayed for the binding affinity, of which four compounds can bind InhA well with KD values ranging from 48.4 to 56.2 µM. Among them, compound 9,222,034 has the best inhibitory activity against InhA enzyme with an IC50 value of 18.05 µM. In addition, the molecular dynamic simulation and binding free energy calculation indicate that the identified compounds bind to InhA with “extended” conformation. Residue energy decomposition shows that residues such as Tyr158, Met161, and Met191 have higher energy contributions in the binding of compounds. By analyzing the binding modes, we found that these compounds can bind to a hydrophobic sub-pocket formed by residues Tyr158, Phe149, Ile215, Leu218, etc., resulting in extensive van der Waals interactions. In summary, this study proposed an efficient strategy for discovering InhA direct inhibitors through ensemble docking-based virtual screening, and finally identified four active compounds with new skeletons, which can provide valuable information for the discovery and optimization of InhA direct inhibitors.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


