Jacob M Remington, Kyle T McKay, Noah B Beckage, Jonathon B Ferrell, Severin T. Schneebeli, Jianing Li
{"title":"GPCRLigNet: rapid screening for GPCR active ligands using machine learning","authors":"Jacob M Remington, Kyle T McKay, Noah B Beckage, Jonathon B Ferrell, Severin T. Schneebeli, Jianing Li","doi":"10.1007/s10822-023-00497-2","DOIUrl":null,"url":null,"abstract":"<div><p>Molecules with bioactivity towards G protein-coupled receptors represent a subset of the vast space of small drug-like molecules. Here, we compare machine learning models, including dilated graph convolutional networks, that conduct binary classification to quickly identify molecules with activity towards G protein-coupled receptors. The models are trained and validated using a large set of over 600,000 active, inactive, and decoy compounds. The best performing machine learning model, dubbed GPCRLigNet, was a surprisingly simple feedforward dense neural network mapping from Morgan fingerprints to activity. Incorporation of GPCRLigNet into a high-throughput virtual screening workflow is demonstrated with molecular docking towards a particular G protein-coupled receptor, the pituitary adenylate cyclase-activating polypeptide receptor type 1. Through rigorous comparison of docking scores for molecules selected with and without using GPCRLigNet, we demonstrate an enrichment of potentially potent molecules using GPCRLigNet. This work provides a proof of principle that GPCRLigNet can effectively hone the chemical search space towards ligands with G protein-coupled receptor activity.</p></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"37 3","pages":"147 - 156"},"PeriodicalIF":3.0000,"publicationDate":"2023-02-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s10822-023-00497-2.pdf","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-023-00497-2","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
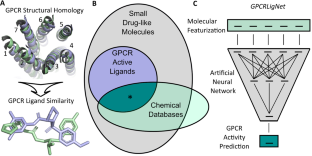
Molecules with bioactivity towards G protein-coupled receptors represent a subset of the vast space of small drug-like molecules. Here, we compare machine learning models, including dilated graph convolutional networks, that conduct binary classification to quickly identify molecules with activity towards G protein-coupled receptors. The models are trained and validated using a large set of over 600,000 active, inactive, and decoy compounds. The best performing machine learning model, dubbed GPCRLigNet, was a surprisingly simple feedforward dense neural network mapping from Morgan fingerprints to activity. Incorporation of GPCRLigNet into a high-throughput virtual screening workflow is demonstrated with molecular docking towards a particular G protein-coupled receptor, the pituitary adenylate cyclase-activating polypeptide receptor type 1. Through rigorous comparison of docking scores for molecules selected with and without using GPCRLigNet, we demonstrate an enrichment of potentially potent molecules using GPCRLigNet. This work provides a proof of principle that GPCRLigNet can effectively hone the chemical search space towards ligands with G protein-coupled receptor activity.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


