Xiao Liu, Lei Zheng, Yalong Cong, Zhihao Gong, Zhixiang Yin, John Z. H. Zhang, Zhirong Liu, Zhaoxi Sun
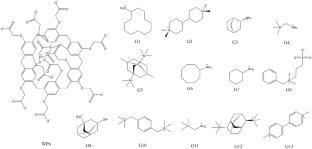
{"title":"Comprehensive evaluation of end-point free energy techniques in carboxylated-pillar[6]arene host–guest binding: II. regression and dielectric constant","authors":"Xiao Liu, Lei Zheng, Yalong Cong, Zhihao Gong, Zhixiang Yin, John Z. H. Zhang, Zhirong Liu, Zhaoxi Sun","doi":"10.1007/s10822-022-00487-w","DOIUrl":null,"url":null,"abstract":"<div><p>End-point free energy calculations as a powerful tool have been widely applied in protein–ligand and protein–protein interactions. It is often recognized that these end-point techniques serve as an option of intermediate accuracy and computational cost compared with more rigorous statistical mechanic models (e.g., alchemical transformation) and coarser molecular docking. However, it is observed that this intermediate level of accuracy does not hold in relatively simple and prototypical host–guest systems. Specifically, in our previous work investigating a set of carboxylated-pillar[6]arene host–guest complexes, end-point methods provide free energy estimates deviating significantly from the experimental reference, and the rank of binding affinities is also incorrectly computed. These observations suggest the unsuitability and inapplicability of standard end-point free energy techniques in host–guest systems, and alteration and development are required to make them practically usable. In this work, we consider two ways to improve the performance of end-point techniques. The first one is the PBSA_E regression that varies the weights of different free energy terms in the end-point calculation procedure, while the second one is considering the interior dielectric constant as an additional variable in the end-point equation. By detailed investigation of the calculation procedure and the simulation outcome, we prove that these two treatments (i.e., regression and dielectric constant) are manipulating the end-point equation in a somehow similar way, i.e., weakening the electrostatic contribution and strengthening the non-polar terms, although there are still many detailed differences between these two methods. With the trained end-point scheme, the RMSE of the computed affinities is improved from the standard ~ 12 kcal/mol to ~ 2.4 kcal/mol, which is comparable to another altered end-point method (ELIE) trained with system-specific data. By tuning PBSA_E weighting factors with the host-specific data, it is possible to further decrease the prediction error to ~ 2.1 kcal/mol. These observations along with the extremely efficient optimized-structure computation procedure suggest the regression (i.e., PBSA_E as well as its GBSA_E extension) as a practically applicable solution that brings end-point methods back into the library of usable tools for host–guest binding. However, the dielectric-constant-variable scheme cannot effectively minimize the experiment-calculation discrepancy for absolute binding affinities, but is able to improve the calculation of affinity ranks. This phenomenon is somehow different from the protein–ligand case and suggests the difference between host–guest and biomacromolecular (protein–ligand and protein–protein) systems. Therefore, the spectrum of tools usable for protein–ligand complexes could be unsuitable for host–guest binding, and numerical validations are necessary to screen out really workable solutions in these ‘prototypical’ situations.\n</p></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"36 12","pages":"879 - 894"},"PeriodicalIF":3.0000,"publicationDate":"2022-11-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"6","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-022-00487-w","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 6
Abstract
End-point free energy calculations as a powerful tool have been widely applied in protein–ligand and protein–protein interactions. It is often recognized that these end-point techniques serve as an option of intermediate accuracy and computational cost compared with more rigorous statistical mechanic models (e.g., alchemical transformation) and coarser molecular docking. However, it is observed that this intermediate level of accuracy does not hold in relatively simple and prototypical host–guest systems. Specifically, in our previous work investigating a set of carboxylated-pillar[6]arene host–guest complexes, end-point methods provide free energy estimates deviating significantly from the experimental reference, and the rank of binding affinities is also incorrectly computed. These observations suggest the unsuitability and inapplicability of standard end-point free energy techniques in host–guest systems, and alteration and development are required to make them practically usable. In this work, we consider two ways to improve the performance of end-point techniques. The first one is the PBSA_E regression that varies the weights of different free energy terms in the end-point calculation procedure, while the second one is considering the interior dielectric constant as an additional variable in the end-point equation. By detailed investigation of the calculation procedure and the simulation outcome, we prove that these two treatments (i.e., regression and dielectric constant) are manipulating the end-point equation in a somehow similar way, i.e., weakening the electrostatic contribution and strengthening the non-polar terms, although there are still many detailed differences between these two methods. With the trained end-point scheme, the RMSE of the computed affinities is improved from the standard ~ 12 kcal/mol to ~ 2.4 kcal/mol, which is comparable to another altered end-point method (ELIE) trained with system-specific data. By tuning PBSA_E weighting factors with the host-specific data, it is possible to further decrease the prediction error to ~ 2.1 kcal/mol. These observations along with the extremely efficient optimized-structure computation procedure suggest the regression (i.e., PBSA_E as well as its GBSA_E extension) as a practically applicable solution that brings end-point methods back into the library of usable tools for host–guest binding. However, the dielectric-constant-variable scheme cannot effectively minimize the experiment-calculation discrepancy for absolute binding affinities, but is able to improve the calculation of affinity ranks. This phenomenon is somehow different from the protein–ligand case and suggests the difference between host–guest and biomacromolecular (protein–ligand and protein–protein) systems. Therefore, the spectrum of tools usable for protein–ligand complexes could be unsuitable for host–guest binding, and numerical validations are necessary to screen out really workable solutions in these ‘prototypical’ situations.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


