The slow but steady rise of binding free energy calculations in drug discovery
IF 3
3区 生物学
Q3 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 6
Abstract
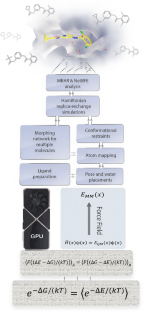
Binding free energy calculations are increasingly used in drug discovery research to predict protein-ligand binding affinities and to prioritize candidate drug molecules accordingly. It has taken decades of collective effort to transform this academic concept into a technology adopted by the pharmaceutical and biotech industry. Having personally witnessed and taken part in this transformation, here I recount the (incomplete) list of problems that had to be solved to make this computational tool practical and suggest areas of future development.
药物发现中结合自由能计算的缓慢而稳定的增长
结合自由能计算越来越多地用于药物发现研究,以预测蛋白质与配体的结合亲和力,并相应地优先考虑候选药物分子。经过数十年的集体努力,将这一学术概念转化为制药和生物技术行业采用的技术。在亲身见证并参与了这一转变之后,我在这里列出了(不完整的)必须解决的问题清单,以使这一计算工具变得实用,并提出了未来发展的领域。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Computer-Aided Molecular Design
生物-计算机:跨学科应用
CiteScore
8.00
自引率
8.60%
发文量
56
审稿时长
3 months
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


