Marianna Stampolaki, Ioannis Stylianakis, Helen I. Zgurskaya, Antonios Kolocouris
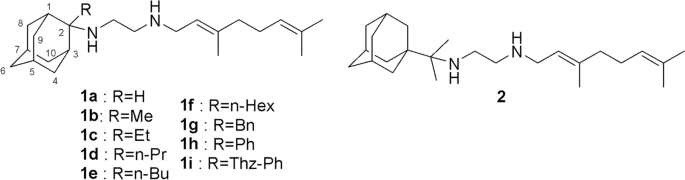
{"title":"Study of SQ109 analogs binding to mycobacterium MmpL3 transporter using MD simulations and alchemical relative binding free energy calculations","authors":"Marianna Stampolaki, Ioannis Stylianakis, Helen I. Zgurskaya, Antonios Kolocouris","doi":"10.1007/s10822-023-00504-6","DOIUrl":null,"url":null,"abstract":"<div><p><i>N</i>-geranyl-<i>N</i>΄-(2-adamantyl)ethane-1,2-diamine (SQ109) is a tuberculosis drug that has high potency against <i>Mycobacterium tuberculosis (Mtb)</i> and may function by blocking cell wall biosynthesis. After the crystal structure of MmpL3 from <i>Mycobacterium smegmatis</i> in complex with SQ109 became available, it was suggested that SQ109 inhibits Mmpl3 mycolic acid transporter. Here, we showed using molecular dynamics (MD) simulations that the binding profile of nine SQ109 analogs with inhibitory potency against Mtb and alkyl or aryl adducts at C-2 or C-1 adamantyl carbon to MmpL3 was consistent with the X-ray structure of MmpL3 – SQ109 complex. We showed that rotation of SQ109 around carbon–carbon bond in the monoprotonated ethylenediamine unit favors two <i>gauche</i> conformations as minima in water and lipophilic solvent using DFT calculations as well as inside the transporter’s binding area using MD simulations. The binding assays in micelles suggested that the binding affinity of the SQ109 analogs was increased for the larger, more hydrophobic adducts, which was consistent with our results from MD simulations of the SQ109 analogues suggesting that sizeable C-2 adamantyl adducts of SQ109 can fill a lipophilic region between Y257, Y646, F260 and F649 in MmpL3. This was confirmed quantitatively by our calculations of the relative binding free energies using the thermodynamic integration coupled with MD simulations method with a mean assigned error of 0.74 kcal mol<sup>−1</sup> compared to the experimental values.</p><h3>Graphical abstract</h3>\n <figure><div><div><div><picture><source><img></source></picture></div></div></div></figure>\n </div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"37 5-6","pages":"245 - 264"},"PeriodicalIF":3.0000,"publicationDate":"2023-05-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s10822-023-00504-6.pdf","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-023-00504-6","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
N-geranyl-N΄-(2-adamantyl)ethane-1,2-diamine (SQ109) is a tuberculosis drug that has high potency against Mycobacterium tuberculosis (Mtb) and may function by blocking cell wall biosynthesis. After the crystal structure of MmpL3 from Mycobacterium smegmatis in complex with SQ109 became available, it was suggested that SQ109 inhibits Mmpl3 mycolic acid transporter. Here, we showed using molecular dynamics (MD) simulations that the binding profile of nine SQ109 analogs with inhibitory potency against Mtb and alkyl or aryl adducts at C-2 or C-1 adamantyl carbon to MmpL3 was consistent with the X-ray structure of MmpL3 – SQ109 complex. We showed that rotation of SQ109 around carbon–carbon bond in the monoprotonated ethylenediamine unit favors two gauche conformations as minima in water and lipophilic solvent using DFT calculations as well as inside the transporter’s binding area using MD simulations. The binding assays in micelles suggested that the binding affinity of the SQ109 analogs was increased for the larger, more hydrophobic adducts, which was consistent with our results from MD simulations of the SQ109 analogues suggesting that sizeable C-2 adamantyl adducts of SQ109 can fill a lipophilic region between Y257, Y646, F260 and F649 in MmpL3. This was confirmed quantitatively by our calculations of the relative binding free energies using the thermodynamic integration coupled with MD simulations method with a mean assigned error of 0.74 kcal mol−1 compared to the experimental values.
n -香叶酰- n -(2-金刚烷基)乙烷-1,2-二胺(SQ109)是一种抗结核分枝杆菌(Mtb)的高效药物,可能通过阻断细胞壁生物合成发挥作用。在获得耻垢分枝杆菌中MmpL3与SQ109复合物的晶体结构后,提示SQ109抑制MmpL3霉菌酸转运体。本研究通过分子动力学(MD)模拟表明,9个具有抑制Mtb和C-2或C-1金刚烷基碳上烷基或芳基加合物的SQ109类似物与MmpL3的结合谱与MmpL3 - SQ109配合物的x射线结构一致。我们发现,在单质子化乙二胺单元中,SQ109围绕碳-碳键的旋转在水和亲脂溶剂中有利于两个间扭构象的最小值,并且在转运体的结合区域内使用MD模拟。胶束结合实验表明,大的、疏水的加合物增加了SQ109类似物的结合亲和力,这与我们在SQ109类似物的MD模拟结果一致,表明SQ109的大的C-2 adamantyl加合物可以填充MmpL3中Y257、Y646、F260和F649之间的亲脂区。我们用热力学积分耦合MD模拟方法计算了相对束缚自由能,与实验值相比,平均分配误差为0.74 kcal mol−1,定量地证实了这一点。图形抽象
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


