Xiaoqiao Li, Ming Cheng, Min Liu, Wenjing Li, Yuchuan Li, Bingyan Cao, Liya Wei, Yuan Ding, Xi Meng, Lele Li, Chunxiu Gong
{"title":"Clinical and genetic characteristics of Cornelia de Lange syndrome in pediatric patients.","authors":"Xiaoqiao Li, Ming Cheng, Min Liu, Wenjing Li, Yuchuan Li, Bingyan Cao, Liya Wei, Yuan Ding, Xi Meng, Lele Li, Chunxiu Gong","doi":"10.1002/ped4.70013","DOIUrl":null,"url":null,"abstract":"<p><strong>Importance: </strong>Cornelia de Lange syndrome (CdLS) is a rare genetic disorder characterized by a spectrum of developmental and physical anomalies. Understanding the clinical and genetic landscape of CdLS in pediatric patients is crucial for improving diagnosis and management.</p><p><strong>Objective: </strong>To investigate the clinical and genetic characteristics of 19 pediatric patients with CdLS in China, with a focus on identifying the association between genetic variants and clinical severity.</p><p><strong>Methods: </strong>We performed whole exome sequencing on 19 patients with CdLS and compared their clinical characteristics based on the presence of null variants.</p><p><strong>Results: </strong>Among the 19 patients, 16 (84.2%) showed global developmental delays and 14 (73.7%) experienced prenatal growth retardation and short stature. Craniofacial anomalies-short noses and anteverted nares were observed in 94.7% (18/19) of patients. Small hands and/or feet were present in 16 patients, skin manifestations (hirsutism or mottled skin) in six, and hearing loss in four. Genetic testing identified 19 variants in <i>NIPBL</i> (78.9%, 15/19), <i>SMC1A</i> (10.5%, 2/19), and <i>RAD21</i> (10.5%, 2/19), including 13 novel variants. <i>NIPBL</i> null variants correlated significantly with more severe growth impairments (<i>P</i> = 0.016) and microcephaly (<i>P</i> = 0.004). Although complete protein function loss often correlated with more severe clinical presentations, no significant difference in clinical scoring was observed (<i>P</i> = 0.600). Three patients treated with recombinant human growth hormone showed heterogeneous responses.</p><p><strong>Interpretation: </strong>This study highlights the clinical heterogeneity of CdLS and suggests a potential link between specific genetic variants and disease severity. These findings warrant further research to optimize treatments and better understand the functional impact of these genetic variants.</p>","PeriodicalId":19992,"journal":{"name":"Pediatric Investigation","volume":"9 3","pages":"293-299"},"PeriodicalIF":2.0000,"publicationDate":"2025-07-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12442446/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pediatric Investigation","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/ped4.70013","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/9/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"PEDIATRICS","Score":null,"Total":0}
引用次数: 0
Abstract
Importance: Cornelia de Lange syndrome (CdLS) is a rare genetic disorder characterized by a spectrum of developmental and physical anomalies. Understanding the clinical and genetic landscape of CdLS in pediatric patients is crucial for improving diagnosis and management.
Objective: To investigate the clinical and genetic characteristics of 19 pediatric patients with CdLS in China, with a focus on identifying the association between genetic variants and clinical severity.
Methods: We performed whole exome sequencing on 19 patients with CdLS and compared their clinical characteristics based on the presence of null variants.
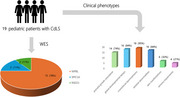
Results: Among the 19 patients, 16 (84.2%) showed global developmental delays and 14 (73.7%) experienced prenatal growth retardation and short stature. Craniofacial anomalies-short noses and anteverted nares were observed in 94.7% (18/19) of patients. Small hands and/or feet were present in 16 patients, skin manifestations (hirsutism or mottled skin) in six, and hearing loss in four. Genetic testing identified 19 variants in NIPBL (78.9%, 15/19), SMC1A (10.5%, 2/19), and RAD21 (10.5%, 2/19), including 13 novel variants. NIPBL null variants correlated significantly with more severe growth impairments (P = 0.016) and microcephaly (P = 0.004). Although complete protein function loss often correlated with more severe clinical presentations, no significant difference in clinical scoring was observed (P = 0.600). Three patients treated with recombinant human growth hormone showed heterogeneous responses.
Interpretation: This study highlights the clinical heterogeneity of CdLS and suggests a potential link between specific genetic variants and disease severity. These findings warrant further research to optimize treatments and better understand the functional impact of these genetic variants.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


