Synergy of advanced machine learning and deep neural networks with consensus molecular docking for virtual screening of anaplastic lymphoma kinase inhibitors
The-Chuong Trinh, Tieu-Long Phan, Van-Thinh To, Thanh-An Pham, Gia-Bao Truong, Lai Hoang Son Le, Xuan-Truc Dinh Tran, Tuyen Ngoc Truong
{"title":"Synergy of advanced machine learning and deep neural networks with consensus molecular docking for virtual screening of anaplastic lymphoma kinase inhibitors","authors":"The-Chuong Trinh, Tieu-Long Phan, Van-Thinh To, Thanh-An Pham, Gia-Bao Truong, Lai Hoang Son Le, Xuan-Truc Dinh Tran, Tuyen Ngoc Truong","doi":"10.1007/s10822-025-00657-6","DOIUrl":null,"url":null,"abstract":"<div><p>This study addresses the urgent need for an AI model to predict Anaplastic Lymphoma Kinase (ALK) inhibitors for Non-Small Cell Lung Cancer treatment, targeting the ALK-positive mutation. With only five Food and Drug Administration approved ALK inhibitors currently available, effective drugs remain in demand. Leveraging machine learning (ML) and deep learning (DL), our research accelerates the precise screening of novel ALK inhibitors using both ligand-based and structure-based approaches. In ligand-based approach, an ensemble voting model comprising three base learners to classify potential ALK inhibitors, achieving promising retrospective validation results. Notably, the ML-based XGBoost algorithm exhibited compelling results with external validation (EV)-f1 score of 0.921, EV-Average Precision (AP) of 0.961, cross-validation (CV)-f1 score of <span>\\(0.888\\pm 0.039\\)</span> and CV-AP of <span>\\(0.939\\pm 0.032\\)</span>. Besides, the DL-based Artificial Neural Network (ANN) model demonstrated comparative performance with EV-f1 score of 0.930, EV-AP of 0.955, CV-f1 score of <span>\\(0.891\\pm 0.037\\)</span> and CV-AP of <span>\\(0.934\\pm 0.040\\)</span>. For structure-based approach, an XGBoost consensus docking model utilized scores from three molecular docking programs (GNINA 1.0, Vina-GPU 2.0, and AutoDock-GPU) as features. Combining these two approaches, we virtually screened 120,571 compounds, identifying three promising ALK inhibitors, CHEMBL1689515, CHEMBL2380351, and CHEMBL102714, that bind to the protein’s pocket and establish hydrophobic contacts in the hinge region through their ketone groups, resembling Alectinib’s interaction. Comparative analysis revealed traditional ML models outperformed Graph Neural Networks (GNN), highlighting the critical role of feature engineering and dataset size importance. The study recommends further in vitro testing to validate the prospective screening performance of these models. A graphical user interface is available at https://huggingface.co/spaces/thechuongtrinh/ALK_inhibitors_classification.</p></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"39 1","pages":""},"PeriodicalIF":3.1000,"publicationDate":"2025-09-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-025-00657-6","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
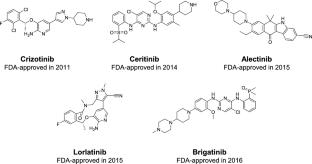
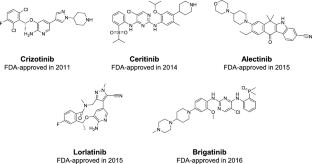
This study addresses the urgent need for an AI model to predict Anaplastic Lymphoma Kinase (ALK) inhibitors for Non-Small Cell Lung Cancer treatment, targeting the ALK-positive mutation. With only five Food and Drug Administration approved ALK inhibitors currently available, effective drugs remain in demand. Leveraging machine learning (ML) and deep learning (DL), our research accelerates the precise screening of novel ALK inhibitors using both ligand-based and structure-based approaches. In ligand-based approach, an ensemble voting model comprising three base learners to classify potential ALK inhibitors, achieving promising retrospective validation results. Notably, the ML-based XGBoost algorithm exhibited compelling results with external validation (EV)-f1 score of 0.921, EV-Average Precision (AP) of 0.961, cross-validation (CV)-f1 score of \(0.888\pm 0.039\) and CV-AP of \(0.939\pm 0.032\). Besides, the DL-based Artificial Neural Network (ANN) model demonstrated comparative performance with EV-f1 score of 0.930, EV-AP of 0.955, CV-f1 score of \(0.891\pm 0.037\) and CV-AP of \(0.934\pm 0.040\). For structure-based approach, an XGBoost consensus docking model utilized scores from three molecular docking programs (GNINA 1.0, Vina-GPU 2.0, and AutoDock-GPU) as features. Combining these two approaches, we virtually screened 120,571 compounds, identifying three promising ALK inhibitors, CHEMBL1689515, CHEMBL2380351, and CHEMBL102714, that bind to the protein’s pocket and establish hydrophobic contacts in the hinge region through their ketone groups, resembling Alectinib’s interaction. Comparative analysis revealed traditional ML models outperformed Graph Neural Networks (GNN), highlighting the critical role of feature engineering and dataset size importance. The study recommends further in vitro testing to validate the prospective screening performance of these models. A graphical user interface is available at https://huggingface.co/spaces/thechuongtrinh/ALK_inhibitors_classification.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


