Yutao Liu, Tinghong Gao, Qingquan Xiao, Yunjun Ruan, Qian Chen, Bei Wang, Jin Huang
{"title":"Generalized modeling of carbon film deposition growth via hybrid MD/MC simulations with machine-learning potentials","authors":"Yutao Liu, Tinghong Gao, Qingquan Xiao, Yunjun Ruan, Qian Chen, Bei Wang, Jin Huang","doi":"10.1038/s41524-025-01781-5","DOIUrl":null,"url":null,"abstract":"<p>Theoretical investigations into the controlled growth of carbon films are essential for guiding the experimental fabrication of carbon-based devices. However, accurately simulating the deposition process remains a significant challenge. In this work, we developed an active learning workflow to construct a machine learning-based neuroevolution potential (NEP) for investigating carbon atoms deposition growth on various substrates. By integrating molecular dynamics and time-stamped force-biased Monte Carlo simulations, we studied the growth of amorphous carbon films on Si(111) and found that deposition energy strongly influenced bonding topology and film morphology. The NEP reliably captured the surface diffusion of carbon atoms, the formation of carbon chains and rings. We revealed a new growth mechanism of adhesion-driven growth at low energies and peening-induced densification at high energies of carbon atoms on Si(111) substrates. To evaluate the transferability of fitting workflow, we extended the NEP to simulate carbon deposition on Cu(111) and Al<sub>2</sub>O<sub>3</sub>(0001) surface. Simulation results demonstrate that the NEP can reproduce the subprocesses of graphene formation during carbon growth on the Cu(111) substrate. In contrast, only disordered carbon chains are observed on the Al<sub>2</sub>O<sub>3</sub>(0001) substrate. This work provides atomistic insights into the growth mechanisms of carbon films on representative substrates and establishes a robust computational framework for synthesis of diverse carbon nanostructures.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"14 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2025-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-025-01781-5","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
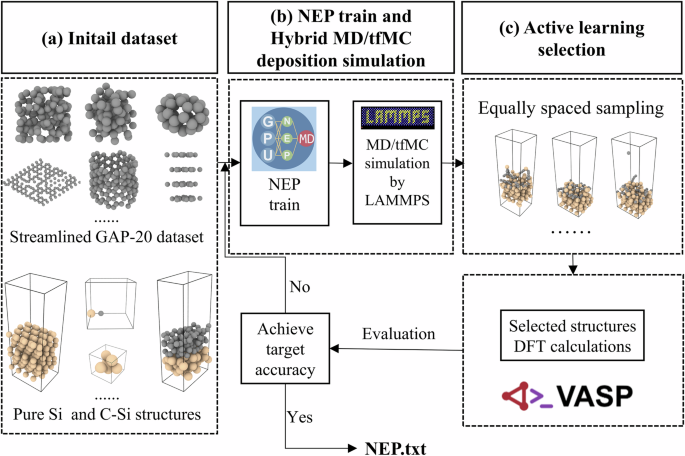
Theoretical investigations into the controlled growth of carbon films are essential for guiding the experimental fabrication of carbon-based devices. However, accurately simulating the deposition process remains a significant challenge. In this work, we developed an active learning workflow to construct a machine learning-based neuroevolution potential (NEP) for investigating carbon atoms deposition growth on various substrates. By integrating molecular dynamics and time-stamped force-biased Monte Carlo simulations, we studied the growth of amorphous carbon films on Si(111) and found that deposition energy strongly influenced bonding topology and film morphology. The NEP reliably captured the surface diffusion of carbon atoms, the formation of carbon chains and rings. We revealed a new growth mechanism of adhesion-driven growth at low energies and peening-induced densification at high energies of carbon atoms on Si(111) substrates. To evaluate the transferability of fitting workflow, we extended the NEP to simulate carbon deposition on Cu(111) and Al2O3(0001) surface. Simulation results demonstrate that the NEP can reproduce the subprocesses of graphene formation during carbon growth on the Cu(111) substrate. In contrast, only disordered carbon chains are observed on the Al2O3(0001) substrate. This work provides atomistic insights into the growth mechanisms of carbon films on representative substrates and establishes a robust computational framework for synthesis of diverse carbon nanostructures.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


