Large-scale genome-wide analyses with proteomics integration reveal novel loci and biological insights into frailty
IF 19.4
Q1 CELL BIOLOGY
引用次数: 0
Abstract
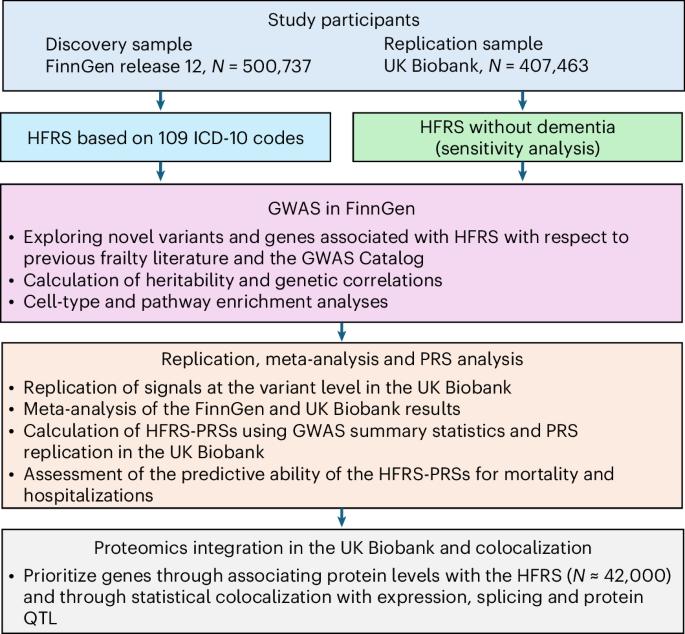
Frailty is a clinically relevant phenotype with notable gaps in our understanding of its etiology. Using the Hospital Frailty Risk Score (HFRS) to define frailty, we performed a genome-wide association study in FinnGen (N = 500,737), replicated the results in the UK Biobank (N = 407,463) and performed a meta-analysis. We prioritized genes through colocalization with expression, splicing and protein quantitative trait loci and proteomics integration. We identified 53 independent lead variants associated with frailty (P < 5 × 10−8), of which 45 were novel and not previously reported in the GWAS Catalog. Replication at the individual variant and polygenic risk score of the HFRS (P = 1.86 × 10−522) levels and meta-analysis largely confirmed the findings. Colocalization analysis supported a causal role for several genes, including CHST9, C6orf106 (ILRUN), KHK, MET, APOE, CGREF1 and PPP6C. Additionally, plasma levels of MET, CGREF1 and APOE were associated with HFRS. Our results reveal new genetic contributions to frailty and shed light on its biological basis. To better understand the etiology of frailty, the authors perform a large genetic study. They identified 45 additional variants and implicated MET, CHST9, ILRUN, APOE, CGREF1 and PPP6C as potential causal genes, linking frailty to immune regulation, metabolism and cellular signaling.
大规模全基因组分析与蛋白质组学整合揭示新的位点和生物学见解脆弱。
虚弱是一种与临床相关的表型,在我们对其病因的理解上存在显著差距。使用医院虚弱风险评分(HFRS)来定义虚弱,我们在FinnGen进行了一项全基因组关联研究(N = 500,737),在UK Biobank (N = 407,463)中重复了结果,并进行了荟萃分析。我们通过表达、剪接和蛋白质数量性状位点的共定位以及蛋白质组学整合来确定基因的优先级。我们确定了53个与虚弱相关的独立铅变异(P -8),其中45个是新的,以前没有在GWAS目录中报道过。在HFRS个体变异和多基因风险评分(P = 1.86 × 10-522)水平上的复制和荟萃分析在很大程度上证实了这一发现。共定位分析支持几个基因的因果作用,包括CHST9, C6orf106 (ILRUN), KHK, MET, APOE, CGREF1和PPP6C。此外,血浆MET、CGREF1和APOE水平与HFRS相关。我们的研究结果揭示了新的遗传因素对脆弱的影响,并阐明了其生物学基础。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


