{"title":"DPmoire: a tool for constructing accurate machine learning force fields in moiré systems","authors":"Jiaxuan Liu, Zhong Fang, Hongming Weng, Quansheng Wu","doi":"10.1038/s41524-025-01740-0","DOIUrl":null,"url":null,"abstract":"<p>In moiré systems, the impact of lattice relaxation on electronic band structures is significant, yet the computational demands of first-principles relaxation are prohibitively high due to the large number of atoms involved. To address this challenge, We introduce a robust methodology for the construction of machine learning potentials specifically tailored for moiré structures and present an open-source software package <b><i>DPmoire</i></b> designed to facilitate this process. Utilizing this package, we have developed machine learning force fields (MLFFs) for MX<sub>2</sub> (M = Mo, W; X = S, Se, Te) materials. Our approach not only streamlines the computational process but also ensures accurate replication of the detailed electronic and structural properties typically observed in density functional theory (DFT) relaxations. The MLFFs were rigorously validated against standard DFT results, confirming their efficacy in capturing the complex interplay of atomic interactions within these layered materials.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"26 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2025-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-025-01740-0","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
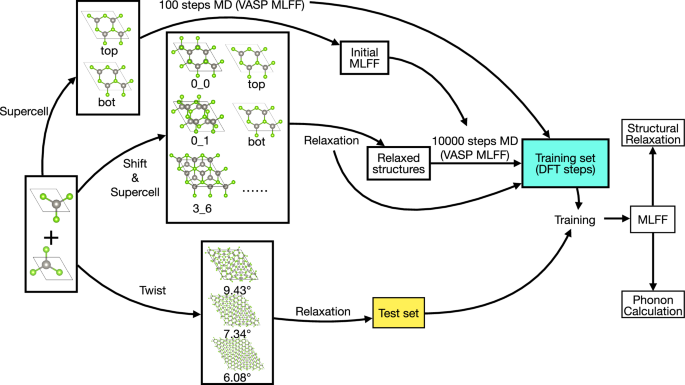
In moiré systems, the impact of lattice relaxation on electronic band structures is significant, yet the computational demands of first-principles relaxation are prohibitively high due to the large number of atoms involved. To address this challenge, We introduce a robust methodology for the construction of machine learning potentials specifically tailored for moiré structures and present an open-source software package DPmoire designed to facilitate this process. Utilizing this package, we have developed machine learning force fields (MLFFs) for MX2 (M = Mo, W; X = S, Se, Te) materials. Our approach not only streamlines the computational process but also ensures accurate replication of the detailed electronic and structural properties typically observed in density functional theory (DFT) relaxations. The MLFFs were rigorously validated against standard DFT results, confirming their efficacy in capturing the complex interplay of atomic interactions within these layered materials.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


