Comprehensive analysis of CNOT3-related neurodevelopmental disorders: phenotypic and genotypic characterization
IF 4.6
2区 生物学
Q2 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
The CCR4-NOT complex, crucial in gene expression regulation, includes CNOT3, a subunit linked to neurodevelopmental disorders when mutated. This study investigates 51 patients from 42 families with heterozygous CNOT3 variants, aiming to expand the understanding of CNOT3-related neurodevelopmental disorders and explore genotype-phenotype correlations. Patients originated from various countries, reflecting the disorder’s global significance. All patients exhibited developmental delays, particularly in the language area. Intellectual disability was found in 87% of patients and was typically mild to moderate. Behavioral issues, including autism spectrum disorders and attention deficits, were common, affecting over half of the patients. Dysmorphic features were highlighted and may help establishing the diagnosis. Epilepsy was uncommon (10%). Twenty-eight novel variants were identified, including missense, nonsense, frameshift, intronic variations and a deletion of 12 exons. Missense variants clustered at the N- and C-terminal regions of the protein, indicating critical functional roles. No clear genotype-phenotype correlation was observed, suggesting that all identified variants resulted in a loss-of-function effect. Finally, this work delineates the clinical and molecular spectrum of CNOT3-related disorders thanks to an in-depth characterization of a large cohort. Further research will be necessary to understand the functional consequences of the variants and enhance patient long-term outcomes.
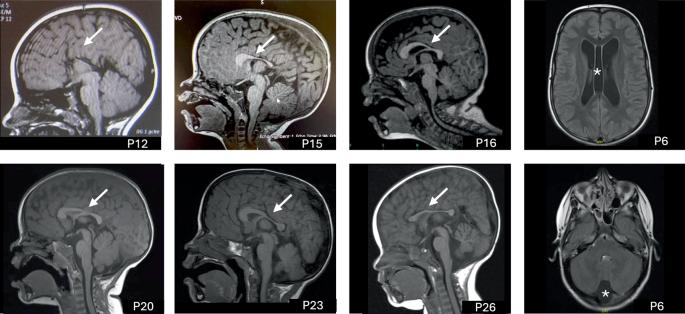
cnot3相关神经发育障碍的综合分析:表型和基因型表征。
CCR4-NOT复合物在基因表达调控中起着至关重要的作用,其中包括CNOT3,这是一种突变时与神经发育障碍相关的亚基。本研究调查了来自42个CNOT3杂合变异家族的51例患者,旨在扩大对CNOT3相关神经发育障碍的认识,并探索基因型-表型相关性。患者来自不同的国家,反映了这种疾病的全球意义。所有患者都表现出发育迟缓,尤其是在语言方面。87%的患者存在智力残疾,通常为轻度至中度。包括自闭症谱系障碍和注意力缺陷在内的行为问题很常见,影响了一半以上的患者。畸形特征突出,可能有助于建立诊断。癫痫不常见(10%)。鉴定出28个新的变异,包括错义、无义、移码、内含子变异和12个外显子的缺失。错义变体聚集在蛋白质的N端和c端区域,表明关键的功能作用。没有观察到明确的基因型-表型相关性,这表明所有已鉴定的变异都会导致功能丧失效应。最后,通过对一个大队列的深入研究,本研究描绘了cnot3相关疾病的临床和分子谱。进一步的研究将是必要的,以了解变异的功能后果和提高患者的长期预后。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

European Journal of Human Genetics
生物-生化与分子生物学
CiteScore
9.90
自引率
5.80%
发文量
216
审稿时长
2 months
期刊介绍:
The European Journal of Human Genetics is the official journal of the European Society of Human Genetics, publishing high-quality, original research papers, short reports and reviews in the rapidly expanding field of human genetics and genomics. It covers molecular, clinical and cytogenetics, interfacing between advanced biomedical research and the clinician, and bridging the great diversity of facilities, resources and viewpoints in the genetics community.
Key areas include:
-Monogenic and multifactorial disorders
-Development and malformation
-Hereditary cancer
-Medical Genomics
-Gene mapping and functional studies
-Genotype-phenotype correlations
-Genetic variation and genome diversity
-Statistical and computational genetics
-Bioinformatics
-Advances in diagnostics
-Therapy and prevention
-Animal models
-Genetic services
-Community genetics
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


