Alireza Khorramfard, Jamshid Pirgazi, Ali Ghanbari Sorkhi
{"title":"Predicting drug protein interactions based on improved support vector data description in unbalanced data.","authors":"Alireza Khorramfard, Jamshid Pirgazi, Ali Ghanbari Sorkhi","doi":"10.34172/bi.30468","DOIUrl":null,"url":null,"abstract":"<p><p></p><p><strong>Introduction: </strong>Predicting drug-protein interactions is critical in drug discovery, but traditional laboratory methods are expensive and time-consuming. Computational approaches, especially those leveraging machine learning, are increasingly popular. This paper introduces VASVDD, a multi-step method to predict drug-protein interactions. First, it extracts features from amino acid sequences in proteins and drug structures. To address the challenge of unbalanced datasets, a Support Vector Data Description (SVDD) approach is employed, outperforming standard techniques like SMOTE and ENN in balancing data. Subsequently, dimensionality reduction using a Variational Autoencoder (VAE) reduces features from 1074 to 32, improving computational efficiency and predictive performance.</p><p><strong>Methods: </strong>The proposed method was evaluated on four datasets related to enzymes, G-protein-coupled receptors, ion channels, and nuclear receptors. Without preprocessing, the Gradient Boosting Classifier showed bias towards the majority class. However, balancing and dimensionality reduction significantly improved accuracy, sensitivity, specificity, and F1 scores. VASVDD demonstrated superior performance compared to other dimensionality reduction methods, such as kernel principal component analysis (kernel PCA) and Principal Component Analysis (PCA), and was validated across multiple classifiers, achieving higher AUROC values than existing techniques.</p><p><strong>Results: </strong>The results highlight VASVDD's effectiveness and generalizability in predicting drug-target interactions. The method outperforms state-of-the-art techniques in terms of accuracy, robustness, and efficiency, making it a promising tool in bioinformatics for drug discovery.</p><p><strong>Conclusion: </strong>The datasets analyzed during the current study are not publicly available but are available from the corresponding author upon reasonable request and source code are available on GitHub: https://github.com/alirezakhorramfard/vasvdd.</p>","PeriodicalId":48614,"journal":{"name":"Bioimpacts","volume":"15 ","pages":"30468"},"PeriodicalIF":2.2000,"publicationDate":"2024-12-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12008248/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioimpacts","FirstCategoryId":"5","ListUrlMain":"https://doi.org/10.34172/bi.30468","RegionNum":4,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 0
Abstract
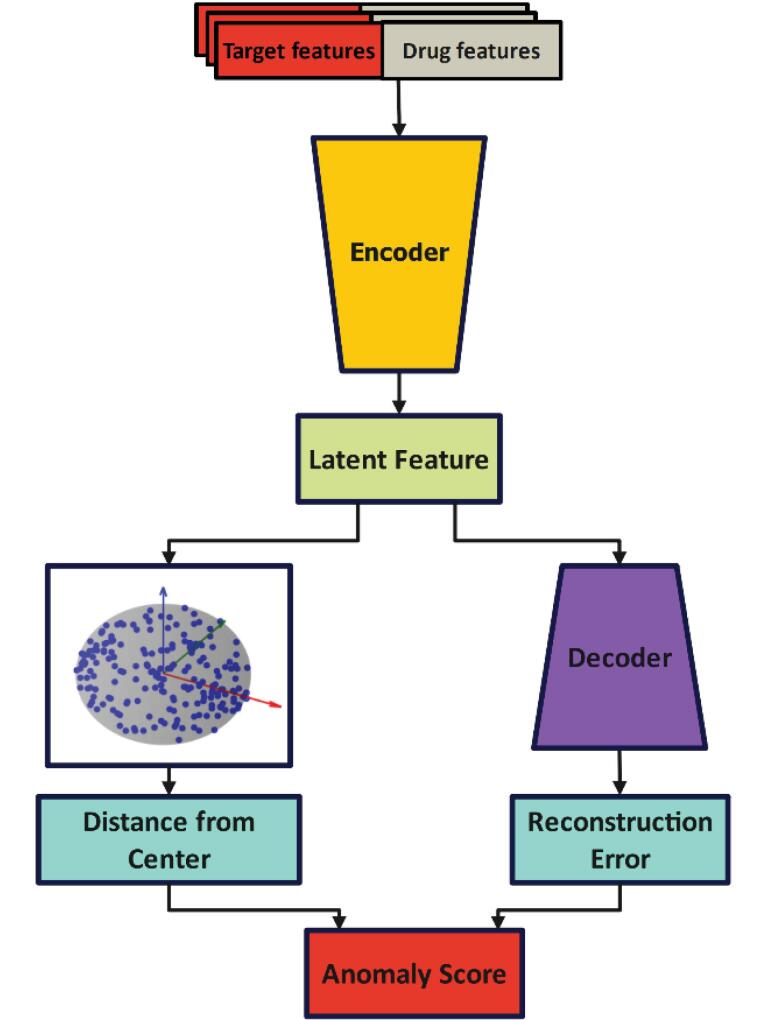
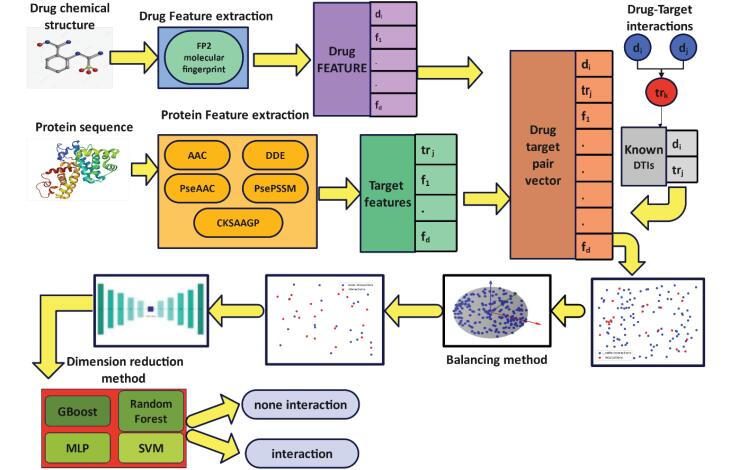
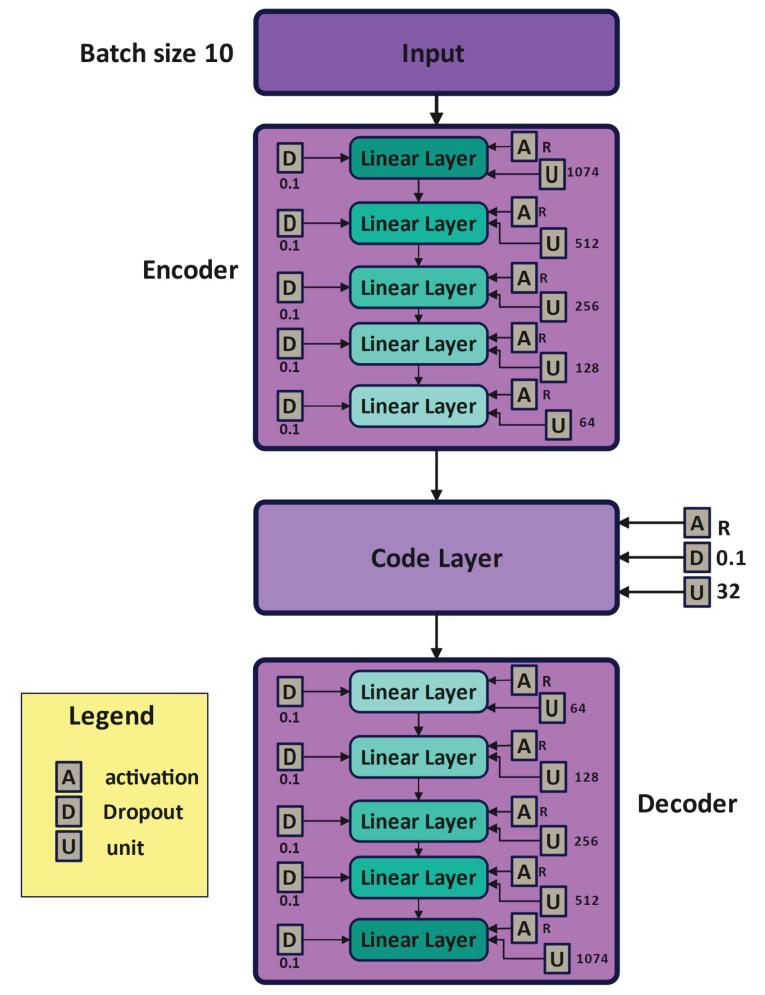
Introduction: Predicting drug-protein interactions is critical in drug discovery, but traditional laboratory methods are expensive and time-consuming. Computational approaches, especially those leveraging machine learning, are increasingly popular. This paper introduces VASVDD, a multi-step method to predict drug-protein interactions. First, it extracts features from amino acid sequences in proteins and drug structures. To address the challenge of unbalanced datasets, a Support Vector Data Description (SVDD) approach is employed, outperforming standard techniques like SMOTE and ENN in balancing data. Subsequently, dimensionality reduction using a Variational Autoencoder (VAE) reduces features from 1074 to 32, improving computational efficiency and predictive performance.
Methods: The proposed method was evaluated on four datasets related to enzymes, G-protein-coupled receptors, ion channels, and nuclear receptors. Without preprocessing, the Gradient Boosting Classifier showed bias towards the majority class. However, balancing and dimensionality reduction significantly improved accuracy, sensitivity, specificity, and F1 scores. VASVDD demonstrated superior performance compared to other dimensionality reduction methods, such as kernel principal component analysis (kernel PCA) and Principal Component Analysis (PCA), and was validated across multiple classifiers, achieving higher AUROC values than existing techniques.
Results: The results highlight VASVDD's effectiveness and generalizability in predicting drug-target interactions. The method outperforms state-of-the-art techniques in terms of accuracy, robustness, and efficiency, making it a promising tool in bioinformatics for drug discovery.
Conclusion: The datasets analyzed during the current study are not publicly available but are available from the corresponding author upon reasonable request and source code are available on GitHub: https://github.com/alirezakhorramfard/vasvdd.
BioimpactsPharmacology, Toxicology and Pharmaceutics-Pharmaceutical Science
CiteScore
4.80
自引率
7.70%
发文量
36
审稿时长
5 weeks
期刊介绍:
BioImpacts (BI) is a peer-reviewed multidisciplinary international journal, covering original research articles, reviews, commentaries, hypotheses, methodologies, and visions/reflections dealing with all aspects of biological and biomedical researches at molecular, cellular, functional and translational dimensions.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


