A new catalytic merit for prediction catalytic potential of 2D materials in LiO2 batteries: Theoretical investigation and experimental identification
IF 9.6
1区 材料科学
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Two-dimensional (2D) materials such as metal chalcogenides have great potential as cathode catalyst materials for lithium oxygen batteries (LOBs) due to their large specific surface area and stable chemical properties. However, thus far, due to the lack of theoretical prediction methods, huge load on catalytic synthesis and performance evaluation is concerned. Herein, we reported a theoretical method for 2D metal chalcogenides as catalysts for LOBs using first principles density functional theory (DFT) calculations. We extracted key parameters that affect the overpotential, including Li–X bond energy (X represents chalcogen elements) and catalyst lattice constant, and theoretically predicted the catalytic performance. The DFT calculation results indicate that MoS2 with appropriate Li–X bond energy and lattice constant has the lowest theoretical overpotential, and its cyclic stability should be higher than other materials under the same conditions. Significantly, we experimentally validated the theoretical predictions presented above. The experimental results shows that pure MoS2 with 2H phase can stably work for more than 220 cycles at a current density of 500 mA/g, and the actual overpotential is lower than other metal chalcogenides. This work provides a swift pathway to accelerate searching high performance catalytic in LOBs.
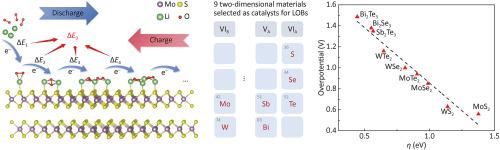

预测二维材料在Li-O2电池中催化电位的新催化优点:理论研究和实验鉴定
金属硫族化合物等二维(2D)材料具有比表面积大、化学性质稳定等优点,作为锂氧电池(lob)正极催化剂材料具有很大的潜力。然而,迄今为止,由于缺乏理论预测方法,对催化合成和性能评价造成了巨大的负担。本文报道了一种基于第一性原理密度泛函理论(DFT)计算的二维金属硫族化合物作为lob催化剂的理论方法。我们提取了影响过电位的关键参数,包括Li-X键能(X代表硫元素)和催化剂晶格常数,并从理论上预测了催化性能。DFT计算结果表明,具有合适的Li-X键能和晶格常数的MoS2具有最低的理论过电位,并且在相同条件下其循环稳定性应高于其他材料。值得注意的是,我们通过实验验证了上述理论预测。实验结果表明,纯2H相MoS2在500 mA/g电流密度下可稳定工作220次以上,实际过电位低于其他金属硫族化合物。这项工作为加速寻找lob中高效催化剂提供了一条快速途径。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Materiomics
Materials Science-Metals and Alloys
CiteScore
14.30
自引率
6.40%
发文量
331
审稿时长
37 days
期刊介绍:
The Journal of Materiomics is a peer-reviewed open-access journal that aims to serve as a forum for the continuous dissemination of research within the field of materials science. It particularly emphasizes systematic studies on the relationships between composition, processing, structure, property, and performance of advanced materials. The journal is supported by the Chinese Ceramic Society and is indexed in SCIE and Scopus. It is commonly referred to as J Materiomics.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


