Energy Resolved Mass Spectrometry Data from Surfaced Induced Dissociation Improves Prediction of Protein Complex Structure
IF 6.7
1区 化学
Q1 CHEMISTRY, ANALYTICAL
引用次数: 0
Abstract
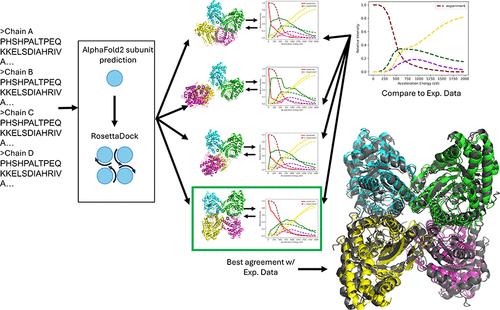
Native Mass Spectrometry (nMS) is a versatile technique for elucidating protein structure. Surface-Induced Dissociation (SID) is an activation method in tandem MS predominantly employed for determining protein complex stoichiometry alongside information about interface strengths. SID-nMS data can be collected over a range of acceleration energies, yielding Energy Resolved Mass Spectrometry (ERMS) data. Previous work demonstrated that the onset and appearance energy from SID-nMS can be used in integrative computational and experimental modeling to guide multimeric structure determination in some cases. However, the appearance energy is a single data point, while the ERMS data provide a full pattern of interface breakage. We hypothesized that incorporation of ERMS data into multimeric protein structure prediction would significantly outperform appearance energy. To test this hypothesis, we generated models of 20 protein complexes with RosettaDock using subunits generated from AlphaFold2. We simulated the ERMS data for each predicted model and rescored based on its agreement to experimental ERMS data. We demonstrated that more accurately predicted models exhibited simulated ERMS data in better agreement with the experimental data. As part of our ERMS-based rescoring, we matched or improved the RMSD of the best scoring model compared to Rosetta in 16 out of 20 cases, with 4 out of 20 cases improving to become a highly accurate (below 5 Å) structure. Finally, we benchmarked our method against our previously published appearance energy-based rescoring and showed improvement in 14 out of 20 cases, with 6 out of 20 becoming a highly accurate (below 5 Å) model. Our method is freely available through Rosetta Commons, with a usage tutorial and test files provided in the Supporting Information.
求助全文
约1分钟内获得全文
求助全文
来源期刊

Analytical Chemistry
化学-分析化学
CiteScore
12.10
自引率
12.20%
发文量
1949
审稿时长
1.4 months
期刊介绍:
Analytical Chemistry, a peer-reviewed research journal, focuses on disseminating new and original knowledge across all branches of analytical chemistry. Fundamental articles may explore general principles of chemical measurement science and need not directly address existing or potential analytical methodology. They can be entirely theoretical or report experimental results. Contributions may cover various phases of analytical operations, including sampling, bioanalysis, electrochemistry, mass spectrometry, microscale and nanoscale systems, environmental analysis, separations, spectroscopy, chemical reactions and selectivity, instrumentation, imaging, surface analysis, and data processing. Papers discussing known analytical methods should present a significant, original application of the method, a notable improvement, or results on an important analyte.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


