Leah R Robinson, Caroline J McDevitt, Molly R Regan, Sophie L Quail, Crista B Wadsworth
{"title":"In vitro evolution of ciprofloxacin resistance in Neisseria commensals and derived mutation population dynamics in natural Neisseria populations.","authors":"Leah R Robinson, Caroline J McDevitt, Molly R Regan, Sophie L Quail, Crista B Wadsworth","doi":"10.1093/femsle/fnae107","DOIUrl":null,"url":null,"abstract":"<p><p>Commensal Neisseria are members of a healthy human oropharyngeal microbiome; however, they also serve as a reservoir of antimicrobial resistance for their pathogenic relatives. Despite their known importance as sources of novel genetic variation for pathogens, we still do not understand the full suite of resistance mutations commensal species can harbor. Here, we use in vitro selection to assess the mutations that emerge in response to ciprofloxacin selection in commensal Neisseria by passaging four replicates of four different species in the presence of a selective antibiotic gradient for 20 days; then categorized derived mutations with whole genome sequencing. Ten out of sixteen selected cells lines across the four species evolved ciprofloxacin resistance (≥1 ug/ml); with resistance-contributing mutations primarily emerging in DNA gyrase subunit A and B (gyrA and gyrB), topoisomerase IV subunits C and E (parC and parE), and the multiple transferable efflux pump repressor (mtrR). Of note, these derived mutations appeared in the same loci responsible for ciprofloxacin-reduced susceptibility in the pathogenic Neisseria, suggesting conserved mechanisms of resistance across the genus. Additionally, we tested for zoliflodacin cross-resistance in evolved strain lines and found 6 lineages with elevated zoliflodacin minimum inhibitory concentrations. Finally, to interrogate the likelihood of experimentally derived mutations emerging and contributing to resistance in natural Neisseria, we used a population-based approach and identified GyrA 91I as a substitution circulating within commensal Neisseria populations and ParC 85C in a single gonococcal isolate. A small cluster of gonococcal isolates shared commensal alleles at parE, suggesting recent cross-species recombination events.</p>","PeriodicalId":12214,"journal":{"name":"Fems Microbiology Letters","volume":" ","pages":""},"PeriodicalIF":2.2000,"publicationDate":"2025-01-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11774118/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Fems Microbiology Letters","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/femsle/fnae107","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
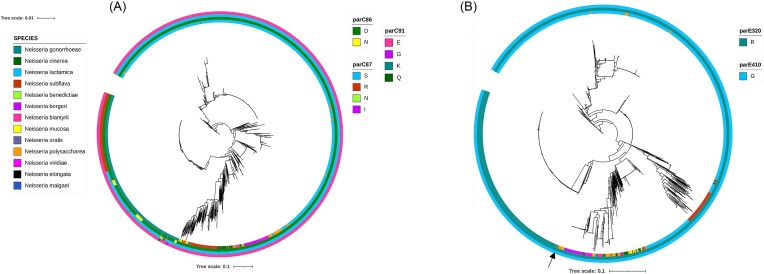
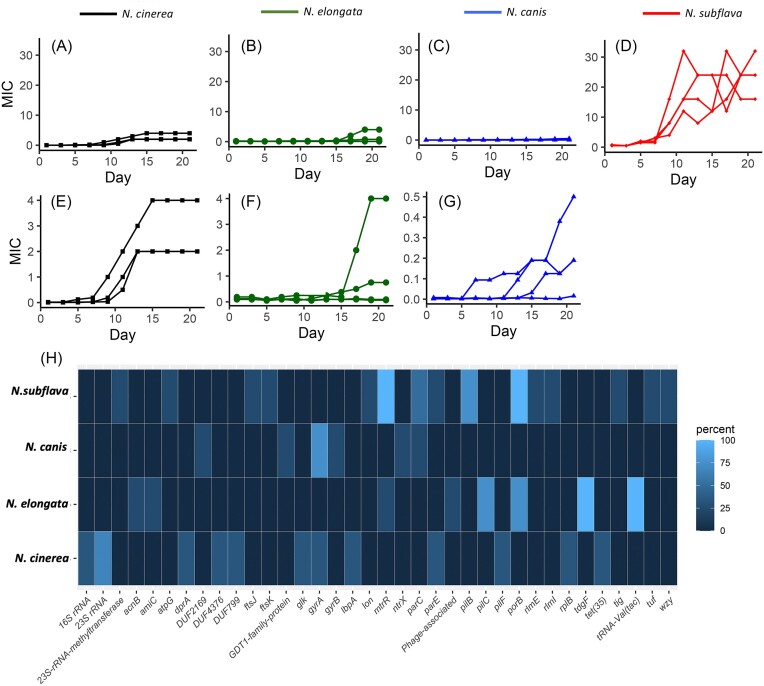
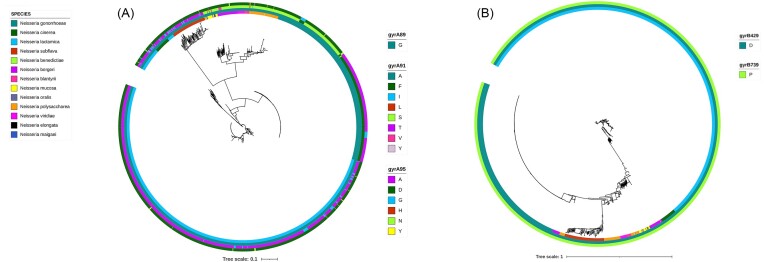
Commensal Neisseria are members of a healthy human oropharyngeal microbiome; however, they also serve as a reservoir of antimicrobial resistance for their pathogenic relatives. Despite their known importance as sources of novel genetic variation for pathogens, we still do not understand the full suite of resistance mutations commensal species can harbor. Here, we use in vitro selection to assess the mutations that emerge in response to ciprofloxacin selection in commensal Neisseria by passaging four replicates of four different species in the presence of a selective antibiotic gradient for 20 days; then categorized derived mutations with whole genome sequencing. Ten out of sixteen selected cells lines across the four species evolved ciprofloxacin resistance (≥1 ug/ml); with resistance-contributing mutations primarily emerging in DNA gyrase subunit A and B (gyrA and gyrB), topoisomerase IV subunits C and E (parC and parE), and the multiple transferable efflux pump repressor (mtrR). Of note, these derived mutations appeared in the same loci responsible for ciprofloxacin-reduced susceptibility in the pathogenic Neisseria, suggesting conserved mechanisms of resistance across the genus. Additionally, we tested for zoliflodacin cross-resistance in evolved strain lines and found 6 lineages with elevated zoliflodacin minimum inhibitory concentrations. Finally, to interrogate the likelihood of experimentally derived mutations emerging and contributing to resistance in natural Neisseria, we used a population-based approach and identified GyrA 91I as a substitution circulating within commensal Neisseria populations and ParC 85C in a single gonococcal isolate. A small cluster of gonococcal isolates shared commensal alleles at parE, suggesting recent cross-species recombination events.
期刊介绍:
FEMS Microbiology Letters gives priority to concise papers that merit rapid publication by virtue of their originality, general interest and contribution to new developments in microbiology. All aspects of microbiology, including virology, are covered.
2019 Impact Factor: 1.987, Journal Citation Reports (Source Clarivate, 2020)
Ranking: 98/135 (Microbiology)
The journal is divided into eight Sections:
Physiology and Biochemistry (including genetics, molecular biology and ‘omic’ studies)
Food Microbiology (from food production and biotechnology to spoilage and food borne pathogens)
Biotechnology and Synthetic Biology
Pathogens and Pathogenicity (including medical, veterinary, plant and insect pathogens – particularly those relating to food security – with the exception of viruses)
Environmental Microbiology (including ecophysiology, ecogenomics and meta-omic studies)
Virology (viruses infecting any organism, including Bacteria and Archaea)
Taxonomy and Systematics (for publication of novel taxa, taxonomic reclassifications and reviews of a taxonomic nature)
Professional Development (including education, training, CPD, research assessment frameworks, research and publication metrics, best-practice, careers and history of microbiology)
If you are unsure which Section is most appropriate for your manuscript, for example in the case of transdisciplinary studies, we recommend that you contact the Editor-In-Chief by email prior to submission. Our scope includes any type of microorganism - all members of the Bacteria and the Archaea and microbial members of the Eukarya (yeasts, filamentous fungi, microbial algae, protozoa, oomycetes, myxomycetes, etc.) as well as all viruses.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


