Pinghui Mo, Yujia Zhang, Zhuoying Zhao, Hanhan Sun, Junhua Li, Dawei Guan, Xi Ding, Xin Zhang, Bo Chen, Mengchao Shi, Duo Zhang, Denghui Lu, Yinan Wang, Jianxing Huang, Fei Liu, Xinyu Li, Mohan Chen, Jun Cheng, Bin Liang, Weinan E, Jiayu Dai, Linfeng Zhang, Han Wang, Jie Liu
{"title":"High-speed and low-power molecular dynamics processing unit (MDPU) with ab initio accuracy","authors":"Pinghui Mo, Yujia Zhang, Zhuoying Zhao, Hanhan Sun, Junhua Li, Dawei Guan, Xi Ding, Xin Zhang, Bo Chen, Mengchao Shi, Duo Zhang, Denghui Lu, Yinan Wang, Jianxing Huang, Fei Liu, Xinyu Li, Mohan Chen, Jun Cheng, Bin Liang, Weinan E, Jiayu Dai, Linfeng Zhang, Han Wang, Jie Liu","doi":"10.1038/s41524-024-01422-3","DOIUrl":null,"url":null,"abstract":"<p>Molecular dynamics (MD) is an indispensable atomistic-scale computational tool widely-used in various disciplines. In the past decades, nearly all ab initio MD and machine-learning MD have been based on the general-purpose central/graphics processing units (CPU/GPU), which are well-known to suffer from their intrinsic “memory wall” and “power wall” bottlenecks. Consequently, nowadays MD calculations with ab initio accuracy are extremely time-consuming and power-consuming, imposing serious restrictions on the MD simulation size and duration. To solve this problem, here we propose a special-purpose MD processing unit (MDPU), which could reduce MD time and power consumption by about 10<sup>3</sup> times (10<sup>9</sup> times) compared to state-of-the-art machine-learning MD (ab initio MD) based on CPU/GPU, while keeping ab initio accuracy. With significantly-enhanced performance, the proposed MDPU may pave a way for the accurate atomistic-scale analysis of large-size and/or long-duration problems which were impossible/impractical to compute before.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"5 1","pages":""},"PeriodicalIF":9.4000,"publicationDate":"2024-11-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01422-3","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
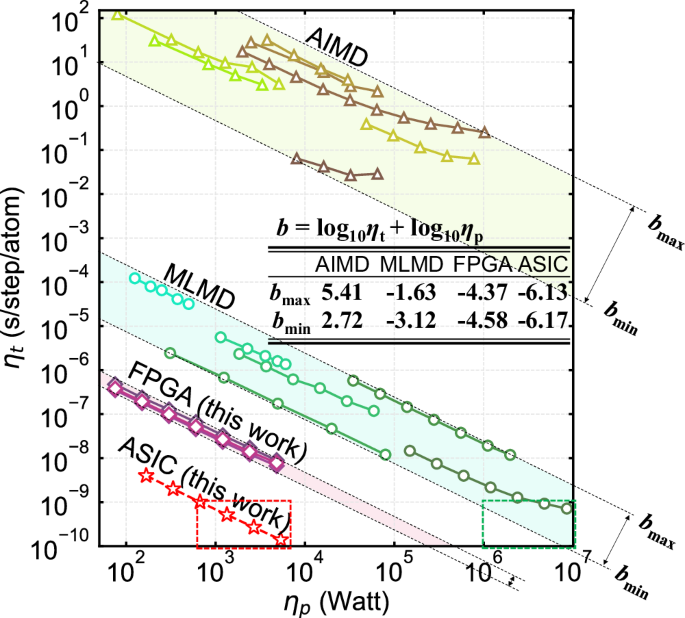
Molecular dynamics (MD) is an indispensable atomistic-scale computational tool widely-used in various disciplines. In the past decades, nearly all ab initio MD and machine-learning MD have been based on the general-purpose central/graphics processing units (CPU/GPU), which are well-known to suffer from their intrinsic “memory wall” and “power wall” bottlenecks. Consequently, nowadays MD calculations with ab initio accuracy are extremely time-consuming and power-consuming, imposing serious restrictions on the MD simulation size and duration. To solve this problem, here we propose a special-purpose MD processing unit (MDPU), which could reduce MD time and power consumption by about 103 times (109 times) compared to state-of-the-art machine-learning MD (ab initio MD) based on CPU/GPU, while keeping ab initio accuracy. With significantly-enhanced performance, the proposed MDPU may pave a way for the accurate atomistic-scale analysis of large-size and/or long-duration problems which were impossible/impractical to compute before.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


