Biallelic and monoallelic variants in EFEMP1 can cause a severe and distinct subtype of heritable connective tissue disorder
IF 3.7
2区 生物学
Q2 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
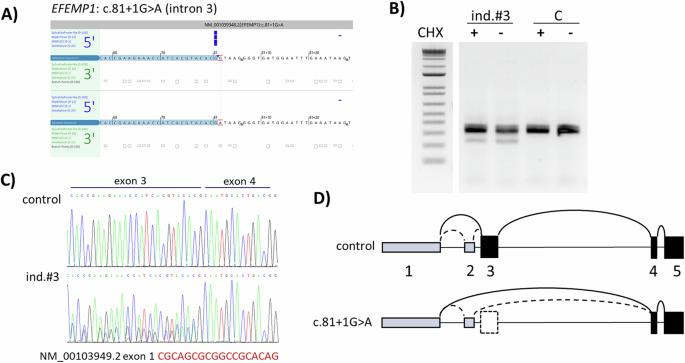
Variants in EFEMP1, encoding Fibulin-3, were previously reported as a rare cause of heritable connective tissue disorder (HCTD) with recurrent hernias and joint hypermobility. We report three new cases with biallelic or monoallelic EFEMP1 variants and severe hernia phenotypes. Two male siblings of 10 and 13 years old presented with marfanoid habitus, recurrent inguinal and umbilical hernias, generalized joint hypermobility, and scoliosis. Parents and halfsiblings reported joint hypermobility and umbilical hernias. The eldest boy died at age 16 from incarcerated gastrointestinal herniation complicated by gastric and bowel necrosis with perforation. Autopsy revealed widespread intestinal diverticula. Immunohistochemistry of skin and fascia tissue did not reveal any abnormalities, including normal staining of elastic fibers. Both siblings harbored compound heterozygous likely pathogenic EFEMP1 variants (c.1320 + 2T > A, p.? and c.698G > A, p.Gly233Asp). An unrelated 58-year-old male had marfanoid features, high myopia, recurrent diaphragmatic and inguinal hernias, and chronic gastrointestinal dilatation with severe malabsorption. Both his dizygotic twin-brother and mother had recurrent hernias and high myopia. This man died at 59 years of age, and autopsy showed extensive diaphragmatic herniation, bowel diverticula, and pulmonary emphysema. A heterozygous EFEMP1 splice-variant (c.81 + 1G > A, p.?) was identified, causing exon skipping leading to a start-loss. Targeted genome reanalysis nor RNA-sequencing revealed a second variant at the other allele. The reported individuals expand the clinical and pathological phenotypes of EFEMP1-related disease, a distinct entity within the spectrum of HCTD. The severe and recurrent hernias, gastrointestinal dilatation, and diverticulosis result in an increased risk for life-threatening complications, demanding early recognition and close monitoring.
EFEMP1 的双拷贝和单拷贝变异可导致一种严重而独特的遗传性结缔组织疾病亚型。
据报道,编码纤维蛋白-3的EFEMP1变异是导致遗传性结缔组织病(HCTD)的一个罕见病因,该病伴有复发性疝气和关节活动过度。我们报告了三个新病例,他们都有双拷贝或单拷贝 EFEMP1 变体和严重的疝气表型。两名分别为 10 岁和 13 岁的男性兄弟姐妹表现为马凡诺形体、复发性腹股沟和脐疝、全身关节活动过度和脊柱侧弯。父母和同父异母的兄弟姐妹都报告了关节活动过度和脐疝。最年长的男孩在16岁时死于嵌顿胃肠疝气,并发胃肠坏死和穿孔。尸体解剖发现了广泛的肠憩室。皮肤和筋膜组织的免疫组化未发现任何异常,包括弹性纤维染色正常。两兄妹均携带可能致病的 EFEMP1 复合杂合子变体(c.1320 + 2T > A, p.? 和 c.698G > A, p.Gly233Asp)。一名没有血缘关系的 58 岁男性具有马凡氏特征、高度近视、复发性膈疝和腹股沟疝、慢性胃肠道扩张和严重吸收不良。他的异卵双胞胎兄弟和母亲都患有复发性疝气和高度近视。这名男子在 59 岁时去世,尸检显示他有广泛的膈疝、肠憩室和肺气肿。经鉴定,他患有杂合子EFEMP1剪接变异体(c.81 + 1G > A, p.?),导致外显子跳转,造成起始缺失。靶向基因组重分析和 RNA 测序发现了另一个等位基因的第二个变异。报告中的患者具有 EFEMP1 相关疾病的临床和病理表型,这是 HCTD 谱系中的一个独特实体。严重和反复发作的疝气、胃肠道扩张和憩室导致危及生命的并发症风险增加,需要早期识别和密切监测。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

European Journal of Human Genetics
生物-生化与分子生物学
CiteScore
9.90
自引率
5.80%
发文量
216
审稿时长
2 months
期刊介绍:
The European Journal of Human Genetics is the official journal of the European Society of Human Genetics, publishing high-quality, original research papers, short reports and reviews in the rapidly expanding field of human genetics and genomics. It covers molecular, clinical and cytogenetics, interfacing between advanced biomedical research and the clinician, and bridging the great diversity of facilities, resources and viewpoints in the genetics community.
Key areas include:
-Monogenic and multifactorial disorders
-Development and malformation
-Hereditary cancer
-Medical Genomics
-Gene mapping and functional studies
-Genotype-phenotype correlations
-Genetic variation and genome diversity
-Statistical and computational genetics
-Bioinformatics
-Advances in diagnostics
-Therapy and prevention
-Animal models
-Genetic services
-Community genetics
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


