Zain Dardas, Dana Marafi, Ruizhi Duan, Jawid M. Fatih, Omnia F. El-Rashidy, Christopher M. Grochowski, Claudia M. B. Carvalho, Shalini N. Jhangiani, Weimin Bi, Haowei Du, Richard A. Gibbs, Jennifer E. Posey, Daniel G. Calame, Maha S. Zaki, James R. Lupski
{"title":"Genomic Balancing Act: deciphering DNA rearrangements in the complex chromosomal aberration involving 5p15.2, 2q31.1, and 18q21.32","authors":"Zain Dardas, Dana Marafi, Ruizhi Duan, Jawid M. Fatih, Omnia F. El-Rashidy, Christopher M. Grochowski, Claudia M. B. Carvalho, Shalini N. Jhangiani, Weimin Bi, Haowei Du, Richard A. Gibbs, Jennifer E. Posey, Daniel G. Calame, Maha S. Zaki, James R. Lupski","doi":"10.1038/s41431-024-01680-1","DOIUrl":null,"url":null,"abstract":"<p>Despite extensive research into the genetic underpinnings of neurodevelopmental disorders (NDD), many clinical cases remain unresolved. We studied a female proband with a NDD, mildly dysmorphic facial features, and brain stem hypoplasia on neuroimaging. Comprehensive genomic analyses revealed a terminal 5p loss and a terminal 18q gain in the proband while a diploid copy number for chromosomes 5 and 18 in both parents. Genomic investigations in the proband identified an unbalanced translocation t(5;18) with additional genetic material from chromosome 2 (2q31.3) inserted at the breakpoint, pointing to a complex chromosomal rearrangement (CCR) involving 5p15.2, 2q31.3, and 18q21.32. Breakpoint junction analyses enabled by long-read genome sequencing unveiled the presence of four distinct junctions in the father, who is a carrier of a balanced CCR. The proband inherited from the father both the abnormal chromosome 5 resulting in segmental aneusomies of chr5 (loss) and chr18 (gain) and a der(2) homologue. Evidences suggest a chromoplexy mechanism for this CCR derivation, involving double-strand breaks (DSBs) repaired by non-homologous end joining (NHEJ) or alternative end joining (alt-EJ). The complexity of the CCR and the segregation of homologues elucidate the genetic model for this family. This study demonstrates the importance of combining multiple genomic technologies to uncover genetic causes of complex neurodevelopmental syndromes and to better understand genetic disease mechanisms.</p>","PeriodicalId":12016,"journal":{"name":"European Journal of Human Genetics","volume":"43 1","pages":""},"PeriodicalIF":3.7000,"publicationDate":"2024-09-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1038/s41431-024-01680-1","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
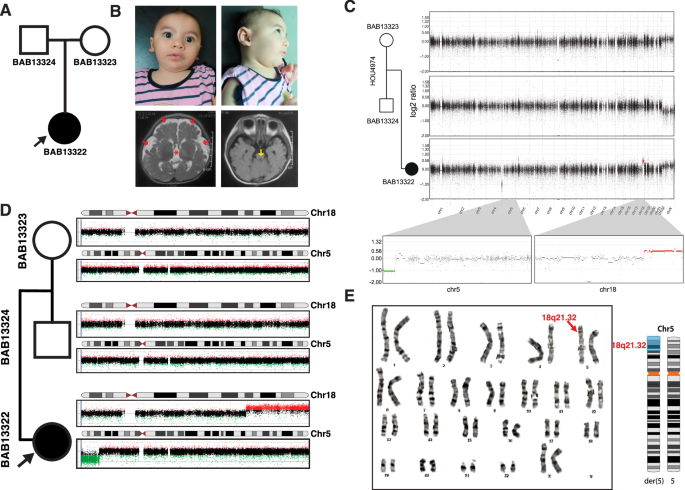
Despite extensive research into the genetic underpinnings of neurodevelopmental disorders (NDD), many clinical cases remain unresolved. We studied a female proband with a NDD, mildly dysmorphic facial features, and brain stem hypoplasia on neuroimaging. Comprehensive genomic analyses revealed a terminal 5p loss and a terminal 18q gain in the proband while a diploid copy number for chromosomes 5 and 18 in both parents. Genomic investigations in the proband identified an unbalanced translocation t(5;18) with additional genetic material from chromosome 2 (2q31.3) inserted at the breakpoint, pointing to a complex chromosomal rearrangement (CCR) involving 5p15.2, 2q31.3, and 18q21.32. Breakpoint junction analyses enabled by long-read genome sequencing unveiled the presence of four distinct junctions in the father, who is a carrier of a balanced CCR. The proband inherited from the father both the abnormal chromosome 5 resulting in segmental aneusomies of chr5 (loss) and chr18 (gain) and a der(2) homologue. Evidences suggest a chromoplexy mechanism for this CCR derivation, involving double-strand breaks (DSBs) repaired by non-homologous end joining (NHEJ) or alternative end joining (alt-EJ). The complexity of the CCR and the segregation of homologues elucidate the genetic model for this family. This study demonstrates the importance of combining multiple genomic technologies to uncover genetic causes of complex neurodevelopmental syndromes and to better understand genetic disease mechanisms.
期刊介绍:
The European Journal of Human Genetics is the official journal of the European Society of Human Genetics, publishing high-quality, original research papers, short reports and reviews in the rapidly expanding field of human genetics and genomics. It covers molecular, clinical and cytogenetics, interfacing between advanced biomedical research and the clinician, and bridging the great diversity of facilities, resources and viewpoints in the genetics community.
Key areas include:
-Monogenic and multifactorial disorders
-Development and malformation
-Hereditary cancer
-Medical Genomics
-Gene mapping and functional studies
-Genotype-phenotype correlations
-Genetic variation and genome diversity
-Statistical and computational genetics
-Bioinformatics
-Advances in diagnostics
-Therapy and prevention
-Animal models
-Genetic services
-Community genetics

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


