{"title":"Adhesion, Stability and Electronic Properties of Ag/SnO2 Interface from First-Principles Calculation","authors":"Yunhui Xu, Jintao Li, Wensong Teng, Defeng Cui, Xiaolong Zhou","doi":"10.1002/crat.202400126","DOIUrl":null,"url":null,"abstract":"<p>The interfacial bonding state between each oxide and the silver matrix in AgCuOIn<sub>2</sub>O<sub>3</sub>SnO<sub>2</sub> electrical contact materials remains unclear. To address this, first-principles calculations using density-functional theory are employed to establish the low-index surfaces of Ag and SnO<sub>2</sub> and perform convergence tests. Computational results reveal that the Ag (111) surface and the SnO<sub>2</sub>(110)-O surface exhibit the highest stability among their respective low-index surfaces. Consequently, these surfaces are chosen to form the interfacial model, and their atomic structure, adhesion work, and interfacial energies are systematically analyzed. The results demonstrate that the stability and interfacial bonding strength of the Ag(111)/SnO<sub>2</sub>(110)-O interface are high, exhibiting metallic properties and strong conductivity. Moreover, at an interface spacing of d<sub>0</sub> = 2.4 Å, the interface stability is optimal. The redistribution of charge at the interface induces significant changes in the local atomic density of states, particularly noticeable in the Ag and O atoms. Additionally, the Ag/SnO<sub>2</sub> interface is predominantly bonded through ionic interactions, contributing to its robust bonding.</p>","PeriodicalId":48935,"journal":{"name":"Crystal Research and Technology","volume":"59 11","pages":""},"PeriodicalIF":1.5000,"publicationDate":"2024-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Crystal Research and Technology","FirstCategoryId":"88","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/crat.202400126","RegionNum":4,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Chemistry","Score":null,"Total":0}
引用次数: 0
Abstract
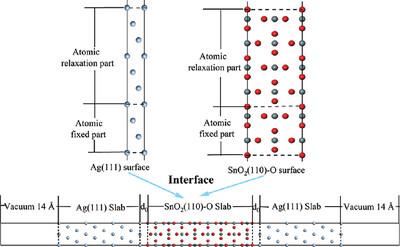
The interfacial bonding state between each oxide and the silver matrix in AgCuOIn2O3SnO2 electrical contact materials remains unclear. To address this, first-principles calculations using density-functional theory are employed to establish the low-index surfaces of Ag and SnO2 and perform convergence tests. Computational results reveal that the Ag (111) surface and the SnO2(110)-O surface exhibit the highest stability among their respective low-index surfaces. Consequently, these surfaces are chosen to form the interfacial model, and their atomic structure, adhesion work, and interfacial energies are systematically analyzed. The results demonstrate that the stability and interfacial bonding strength of the Ag(111)/SnO2(110)-O interface are high, exhibiting metallic properties and strong conductivity. Moreover, at an interface spacing of d0 = 2.4 Å, the interface stability is optimal. The redistribution of charge at the interface induces significant changes in the local atomic density of states, particularly noticeable in the Ag and O atoms. Additionally, the Ag/SnO2 interface is predominantly bonded through ionic interactions, contributing to its robust bonding.
期刊介绍:
The journal Crystal Research and Technology is a pure online Journal (since 2012).
Crystal Research and Technology is an international journal examining all aspects of research within experimental, industrial, and theoretical crystallography. The journal covers the relevant aspects of
-crystal growth techniques and phenomena (including bulk growth, thin films)
-modern crystalline materials (e.g. smart materials, nanocrystals, quasicrystals, liquid crystals)
-industrial crystallisation
-application of crystals in materials science, electronics, data storage, and optics
-experimental, simulation and theoretical studies of the structural properties of crystals
-crystallographic computing

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


